



Abstract
The low temperature diffusivity of nanoscale crystal defects, where quantum mechanical fluctuations are known to play a crucial role, are essential to interpret observations of irradiated microstructures conducted at cryogenic temperatures. Using density functional theory calculations, quantum heat bath molecular dynamics and open quantum systems theory, we evaluate the low temperature diffusivity of self-interstitial atom clusters in tungsten valid down to temperatures of 1 K. Due to an exceptionally low defect migration barrier, our results show that interstitial defects exhibit very high diffusivity of order  over the entire range of temperatures investigated.
over the entire range of temperatures investigated.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Studies of irradiated materials are often conducted at cryogenic temperatures to simplify the spectrum of processes participating in microstructure evolution, allowing valuable information to be extracted through resistivity recovery analysis [1–3] or, more recently, through direct transmission electron microscope observations [4–6]. However, the interpretation of such experiments requires accurate mobility laws for irradiation defects that account for non-classical thermal fluctuations below the Debye temperature  .
.
In this paper we provide a predictive calculation of the mobility of  self-interstitial atom (SIA) clusters in body centered cubic tungsten [7–16], defects of great interest to fission and fusion reactor materials development, valid for all temperatures above approximately 1 K. We find that even at very low temperatures the diffusivity remains very high (of the order of
self-interstitial atom (SIA) clusters in body centered cubic tungsten [7–16], defects of great interest to fission and fusion reactor materials development, valid for all temperatures above approximately 1 K. We find that even at very low temperatures the diffusivity remains very high (of the order of  s−1) and is effectively constant below 50 K. This very high diffusivity is consistent with experimental observations [1, 6, 17], showing that such defects remain mobile at all observation temperatures, down to 4.2 K.
s−1) and is effectively constant below 50 K. This very high diffusivity is consistent with experimental observations [1, 6, 17], showing that such defects remain mobile at all observation temperatures, down to 4.2 K.
The influence of non-classical thermal fluctuations can be broadly classified into three regimes [19, 20], illustrated in figure 1(a). Below around a third of the Debye temperature  , zero-point motion causes thermal vibrations to be of far greater magnitude than their classical expectation value [21], leading to an effective renormalisation of the classical system temperature, and stimulating stochastic motion of defects which in the absence of tunnelling is fundamentally similar to their classical stochastic thermal motion at high temperature. Below a certain crossover temperature
, zero-point motion causes thermal vibrations to be of far greater magnitude than their classical expectation value [21], leading to an effective renormalisation of the classical system temperature, and stimulating stochastic motion of defects which in the absence of tunnelling is fundamentally similar to their classical stochastic thermal motion at high temperature. Below a certain crossover temperature  , incoherent phonon-assisted tunnelling phenomena causes non-classical trajectories at the peak of the migration barrier to contribute significantly to the barrier crossing rate [19]. Finally, below some very small decoherence temperature
, incoherent phonon-assisted tunnelling phenomena causes non-classical trajectories at the peak of the migration barrier to contribute significantly to the barrier crossing rate [19]. Finally, below some very small decoherence temperature  [22] the system can in principle coherently tunnel between vibrational ground states [23]. The latter becomes the dominant migration mechanism as the system approaches absolute zero.
[22] the system can in principle coherently tunnel between vibrational ground states [23]. The latter becomes the dominant migration mechanism as the system approaches absolute zero.

Figure 1. Above: illustration of the temperature regimes as discussed in the main text, with values appropriate for a crowdion defect in tungsten. Experimentally accessible temperatures are shaded in grey. Below: calculations of the migration barrier for a crowdion defect in tungsten, evaluated with an empirical EAM potential [18] using the drag method and in DFT calculations using the NEB method.
Download figure:
Standard image High-resolution imageIn the majority of applications [19], non-classical thermalisation techniques are used to treat very light atoms such as hydrogen, where the crossover temperature  is comparable or even larger than
is comparable or even larger than  , meaning that the central transition of interest is the onset of phonon assisted tunnelling phenomena to the transition rate, where quantum heat bath techniques [24], which capture only the nonclassical thermal vibrations, are very difficult or impossible to apply. In constrast, this work is concerned with defects in heavy crystalline tungsten (
, meaning that the central transition of interest is the onset of phonon assisted tunnelling phenomena to the transition rate, where quantum heat bath techniques [24], which capture only the nonclassical thermal vibrations, are very difficult or impossible to apply. In constrast, this work is concerned with defects in heavy crystalline tungsten ( ) where we show that
) where we show that  dependent on the defect character. As a result, the central transition of interest is the rising influence of zero-point motion with decreasing temperature, which is able to be treated by the quantum heat bath technique.
dependent on the defect character. As a result, the central transition of interest is the rising influence of zero-point motion with decreasing temperature, which is able to be treated by the quantum heat bath technique.
Using density functional theory (DFT) calculations, we find an extremely small single SIA crowdion [7] migration barrier of 2 meV (figure 1), which has an associated crossover temperature  of around 0.6 K. Similar calculations for a vacancy defect, which has a migration barrier of 1.78 eV [18], yields a higher crossover temperature of 7 K, though due to the large migration barrier the vacancy hopping rate will be negligible at all temperatures where quantum fluctuations have a large influence.
of around 0.6 K. Similar calculations for a vacancy defect, which has a migration barrier of 1.78 eV [18], yields a higher crossover temperature of 7 K, though due to the large migration barrier the vacancy hopping rate will be negligible at all temperatures where quantum fluctuations have a large influence.
In this paper we focus on the highly mobile interstitial defects which dominate the kinetics of post irradiation annealing, meaning we can ignore all tunnelling phenomena above 1 K for interstitial defects and consider only the effect of zero-point fluctuations through the use of quantum heat bath molecular dynamics [24], which captures the quantum distribution of thermal vibrations. The results of our simulations are well described by a theoretical expression derived through the use of open quantum systems (OQS) theory [23] and previous work on the coupling of defects and thermal vibrations [8, 16], which we then generalize to larger SIA clusters.
Static calculation of the crowdion migration barrier
To accurately determine the migration barrier for a crowdion defect in tungsten, we performed ab initio calculations using the VASP package [25, 26], with the AM05 [27] pseudopotential including 12 valence electrons. The simulation supercell contained 129 atoms, with 5 × 5 × 5 gamma-point centered k-point grid and a plane-wave energy cutoff of 450 eV. The ion positions and simulation box shape of the 111 crowdion and 111 dumbbell structures were fully relaxed with energy and force convergence at  eV and
eV and  eV
eV  . To determine the energy barrier the nudged elastic band method [28] calculations were performed using seven images between the ground state and saddle point. After completing the calculations, elastic corrections, as proposed and implemented by Varvenne et al [29], were applied to eliminate the effect of elastic periodic image interactions on the system energy. The results of these calculations are shown in figure 1(b), along with similar calculations [30] performed in a supercell of 2001 atoms using an embedded atom method (EAM) interatomic potential [31] developed by Marinica et al [18], which we also use in our molecular dynamics simulations. The EAM calculations, in good agreement with other available interatomic potentials [7], predict a very small crowdion migration barrier of 17.2 meV. This is consistent with classical molecular dynamics simulations, performed in this work and elsewhere [7, 8], which find, in agreement with experiment [14], that crowdions execute free Brownian motion above 150 K, as the migration barrier is overwhelmed by thermal agitations. However, as can be seen in figure 1(b), our DFT calculations yield an even smaller migration barrier of 2.07 meV, in agreement with previous ab initio based approaches to model this defect [32]. As we will see, this extremely small magnitude of the migration barrier has profound implications for the nature of the low temperature defect motion.
. To determine the energy barrier the nudged elastic band method [28] calculations were performed using seven images between the ground state and saddle point. After completing the calculations, elastic corrections, as proposed and implemented by Varvenne et al [29], were applied to eliminate the effect of elastic periodic image interactions on the system energy. The results of these calculations are shown in figure 1(b), along with similar calculations [30] performed in a supercell of 2001 atoms using an embedded atom method (EAM) interatomic potential [31] developed by Marinica et al [18], which we also use in our molecular dynamics simulations. The EAM calculations, in good agreement with other available interatomic potentials [7], predict a very small crowdion migration barrier of 17.2 meV. This is consistent with classical molecular dynamics simulations, performed in this work and elsewhere [7, 8], which find, in agreement with experiment [14], that crowdions execute free Brownian motion above 150 K, as the migration barrier is overwhelmed by thermal agitations. However, as can be seen in figure 1(b), our DFT calculations yield an even smaller migration barrier of 2.07 meV, in agreement with previous ab initio based approaches to model this defect [32]. As we will see, this extremely small magnitude of the migration barrier has profound implications for the nature of the low temperature defect motion.
As mentioned above, below a crossover temperature  , non-classical phonon assisted tunnelling trajectories emerge as stationary action solutions in an imaginary time path integral evaluation of the defect density matrix [19].
, non-classical phonon assisted tunnelling trajectories emerge as stationary action solutions in an imaginary time path integral evaluation of the defect density matrix [19].  is given by
is given by  , where
, where  is the unstable frequency at the top of the migration barrier. Using
is the unstable frequency at the top of the migration barrier. Using  extracted from our EAM calculations for a crowdion in tungsten gives
extracted from our EAM calculations for a crowdion in tungsten gives  K, whilst using the migration barrier found in DFT calculations gives
K, whilst using the migration barrier found in DFT calculations gives  K. This very low crossover temperature allows us to neglect all tunnelings phenomena above ∼1 K and focus only on the effect of quantum zero-point fluctuations on the thermally activated defect motion. In the supplementary material (available online at stacks.iop.org/NJP/19/073024/mmedia) we also provide an estimate of the decoherence temperature, finding
K. This very low crossover temperature allows us to neglect all tunnelings phenomena above ∼1 K and focus only on the effect of quantum zero-point fluctuations on the thermally activated defect motion. In the supplementary material (available online at stacks.iop.org/NJP/19/073024/mmedia) we also provide an estimate of the decoherence temperature, finding  K, as expected due to tungsten's large atomic mass. As existing empirical potentials cannot capture the extremely low crowdion migration barrier found in our DFT simulations, the observed defect diffusivities found in our molecular dynamics simulations must be modified to account for the discrepancy. We return to this point below, when developing our theoretical treatment of the low temperature diffusion.
K, as expected due to tungsten's large atomic mass. As existing empirical potentials cannot capture the extremely low crowdion migration barrier found in our DFT simulations, the observed defect diffusivities found in our molecular dynamics simulations must be modified to account for the discrepancy. We return to this point below, when developing our theoretical treatment of the low temperature diffusion.
Quantum heat bath molecular dynamics
As tunnelling phenomena can be neglected for system temperatures above  for the SIA clusters considered here, semi-classical simulations, which only retain quantum zero-point motion, are adequate to model the low temperature diffusion. To this end, we have implemented the colored noise quantum heat bath proposed by Barrat and Rodney [24] (see supplementary material) in the MD and spin-lattice dynamics program SPILADY [33]. The quantum heat bath technique is an approximate method to capture the quantum lattice vibrations of solids, which cannot treat systems with pathologically strong anharmonicity [34–36] as zero-point energy is erroneously distributed between all the vibrational modes. However, when parametrised with care, the method is in principle able to treat realistic potentials [24, 37], providing the heat bath friction constant is sufficiently high to prevent zero-point energy leakage but sufficiently weak to allow correct anharmonic behaviour. Whilst this is not in general possible for all systems [37], the strength of atomic bonding in tungsten allows this to be achieved with a parametrisation similar to that used in previous studies [24] (see supplementary material).
for the SIA clusters considered here, semi-classical simulations, which only retain quantum zero-point motion, are adequate to model the low temperature diffusion. To this end, we have implemented the colored noise quantum heat bath proposed by Barrat and Rodney [24] (see supplementary material) in the MD and spin-lattice dynamics program SPILADY [33]. The quantum heat bath technique is an approximate method to capture the quantum lattice vibrations of solids, which cannot treat systems with pathologically strong anharmonicity [34–36] as zero-point energy is erroneously distributed between all the vibrational modes. However, when parametrised with care, the method is in principle able to treat realistic potentials [24, 37], providing the heat bath friction constant is sufficiently high to prevent zero-point energy leakage but sufficiently weak to allow correct anharmonic behaviour. Whilst this is not in general possible for all systems [37], the strength of atomic bonding in tungsten allows this to be achieved with a parametrisation similar to that used in previous studies [24] (see supplementary material).
The simulation supercell contained 16001 atoms, with supercell vectors of ![$20a[001],20a[010]$](https://content.cld.iop.org/journals/1367-2630/19/7/073024/revision2/njpaa78eaieqn25.gif) and
and ![$20a[001]$](https://content.cld.iop.org/journals/1367-2630/19/7/073024/revision2/njpaa78eaieqn26.gif) . The crowdion configuration was structurally relaxed with a conjugate gradients minimization and then thermalized with either a quantum or classical heat bath for 0.1 ns. Simulation trajectories were obtained at ten temperatures between 10 and 200 K, at intervals of 10 K between 10 and 80 K, with longer trajectories broken into four independently thermalized segments. The quantum and classical heat bath trajectories were all 4 ns in length, yielding robust statistical data on the defect diffusivity. The defect position was extracted by first time-averaging over a period of ∼0.05 ps, applying a high-pass energy filter to reveal the defect core, then taking the center of mass of the remaining atoms. At lower temperatures, the defect trajectory is characterized by isolated stochastic jumps; in this case, the diffusivity is given by
. The crowdion configuration was structurally relaxed with a conjugate gradients minimization and then thermalized with either a quantum or classical heat bath for 0.1 ns. Simulation trajectories were obtained at ten temperatures between 10 and 200 K, at intervals of 10 K between 10 and 80 K, with longer trajectories broken into four independently thermalized segments. The quantum and classical heat bath trajectories were all 4 ns in length, yielding robust statistical data on the defect diffusivity. The defect position was extracted by first time-averaging over a period of ∼0.05 ps, applying a high-pass energy filter to reveal the defect core, then taking the center of mass of the remaining atoms. At lower temperatures, the defect trajectory is characterized by isolated stochastic jumps; in this case, the diffusivity is given by  , where
, where  is the number of defect jumps,
is the number of defect jumps,  the total simulation time and b the jump length between potential minima. At higher temperatures, typically above 150 K for the simulations presented here, the defect trajectory exhibits a continuous stochastic motion and the diffusivity
the total simulation time and b the jump length between potential minima. At higher temperatures, typically above 150 K for the simulations presented here, the defect trajectory exhibits a continuous stochastic motion and the diffusivity  is calculated through analysis of the mean squared displacement [8, 11].
is calculated through analysis of the mean squared displacement [8, 11].
The simulated defect diffusivities are displayed on Arrhenius plots  in figure 2. The diffusivities extracted from classical heat bath molecular dynamics clearly exhibits the classical Arrhenius relationship
in figure 2. The diffusivities extracted from classical heat bath molecular dynamics clearly exhibits the classical Arrhenius relationship  below 100 K, with an activation energy
below 100 K, with an activation energy  of 0.018 eV, in very good agreement with the value 0.0172 eV extracted from static calculations using the same EAM potential. From previous studies of crowdion motion in bcc metals, it is known that the Arrhenius behavior breaks down at temperatures above 150 K; at high temperature the diffusivity is that of a freely diffusing particle,
of 0.018 eV, in very good agreement with the value 0.0172 eV extracted from static calculations using the same EAM potential. From previous studies of crowdion motion in bcc metals, it is known that the Arrhenius behavior breaks down at temperatures above 150 K; at high temperature the diffusivity is that of a freely diffusing particle,  , where γ is the frictional coupling between the defect and thermal vibrations [7, 8, 16]. However, in contrast to purely classical simulations, the diffusivities extracted from quantum heat bath molecular dynamics deviate significantly from the classical law at low temperature, indicating the influence of quantum zero-point motion on defect migration.
, where γ is the frictional coupling between the defect and thermal vibrations [7, 8, 16]. However, in contrast to purely classical simulations, the diffusivities extracted from quantum heat bath molecular dynamics deviate significantly from the classical law at low temperature, indicating the influence of quantum zero-point motion on defect migration.

{kind=link}
Figure 2. The results from classical and quantum heat bath simulations of crowdion diffusion in tungsten, along with theoretical predictions using our diffusivity derived from open quantum systems (OQS) theory,  , using the migration barrier found using the EAM potential and in DFT calculations. We also plot the classical diffusivity
, using the migration barrier found using the EAM potential and in DFT calculations. We also plot the classical diffusivity  calculated using equation (5) but setting
calculated using equation (5) but setting  .
.
Download figure:
Standard image High-resolution image{kind=link}
Theoretical treatment of low temperature defect diffusion
We have seen that due to the very low migration barriers found in static calculations, we are able to neglect all tunneling phenomena for system temperatures above 1 K, allowing the use of semiclassical theories which retain only quantum expectation values in the vibrational spectrum [23]. In previous work, a theoretical and numerical scheme was developed to analyze the thermal vibrations surrounding migrating crystal defects, initially applied to study the phonon drag force [8, 16]. The method uses a defect migration pathway as calculated from a barrier climbing calculations, defining the defect position coordinate r to be the reaction coordinate along this pathway. By considering possible structural variations along the migration pathway ( ) and perpendicular to the migration pathway (
) and perpendicular to the migration pathway ( ) one can show that, as defect motion is not in general an eigenmode of the host crystal [16, 38], the potential energy V can be expanded as
) one can show that, as defect motion is not in general an eigenmode of the host crystal [16, 38], the potential energy V can be expanded as
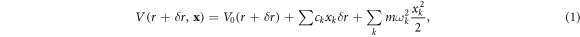
where  is the periodic migration potential shown in figure 1(b). The vibrational frequencies
is the periodic migration potential shown in figure 1(b). The vibrational frequencies  and coupling constants
and coupling constants  are related to the Hessian matrix of second derivatives [8, 16]. The coupling constants
are related to the Hessian matrix of second derivatives [8, 16]. The coupling constants  allow the defect system to be thermalized through interaction with the surrounding thermal vibrations, giving rise (in a classical picture) to a dissipative drag force
allow the defect system to be thermalized through interaction with the surrounding thermal vibrations, giving rise (in a classical picture) to a dissipative drag force  and a fluctuating thermal force of square magnitude
and a fluctuating thermal force of square magnitude  , where the frictional coupling γ can be expressed [8, 16] as a function of the
, where the frictional coupling γ can be expressed [8, 16] as a function of the  and
and  . We have evaluated the
. We have evaluated the  for various defects in bcc metals [8, 16], including the crowdion defect considered here; explicit expressions for
for various defects in bcc metals [8, 16], including the crowdion defect considered here; explicit expressions for  ,
,  and γ are given in the supplementary material.
and γ are given in the supplementary material.
To derive an effective migration rate in the temperature regime of interest, we note that equation (1) is the widely studied 'oscillator bath' Hamiltonian used in the theory of OQS [23], which aims to integrate away the vibrational degrees of freedom  to leave an effective distribution function for the defect density matrix, which then yields a Fokker–Planck equation for the Wigner quasi-probability function
to leave an effective distribution function for the defect density matrix, which then yields a Fokker–Planck equation for the Wigner quasi-probability function  [19]. Expanding the potential around r = 0 to obtain
[19]. Expanding the potential around r = 0 to obtain  , a standard result is that
, a standard result is that  follows the approximate evolution equation [23]
follows the approximate evolution equation [23]

where explicit expressions for D and  are given in the supplementary material. The evolution equation (2) has a steady state distribution
are given in the supplementary material. The evolution equation (2) has a steady state distribution ![${W}_{\infty }(\delta r,{p}_{r})={Z}^{-1}\exp (-[V(\delta r)+{p}_{r}^{2}/2m]\gamma /D)$](https://content.cld.iop.org/journals/1367-2630/19/7/073024/revision2/njpaa78eaieqn56.gif) , that clearly resembles a classical Gibbs distribution with an effective temperature (see supplementary material)
, that clearly resembles a classical Gibbs distribution with an effective temperature (see supplementary material)
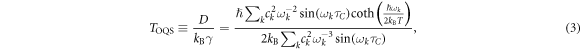
where  ps is the calculated correlation time of the thermal force acting on the defect [8]. It is straightforward to show that the OQS temperature
ps is the calculated correlation time of the thermal force acting on the defect [8]. It is straightforward to show that the OQS temperature  at high temperature, but at low temperature
at high temperature, but at low temperature  tends to a constant value due to zero-point fluctuations. In the inset of figure 2 we compare
tends to a constant value due to zero-point fluctuations. In the inset of figure 2 we compare  to the effective 'classical' temperature of a solid state system with normal modes
to the effective 'classical' temperature of a solid state system with normal modes  [21]
[21]
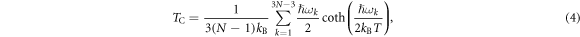
which can also be evaluated numerically through Maxwellian analysis of the atomic velocities to give  . We see that both effective temperatures show a very similar behavior, with
. We see that both effective temperatures show a very similar behavior, with  tending to a slightly lower temperature than
tending to a slightly lower temperature than  . Equations (2) and (3) show that, in the approximation of a harmonic potential well, the coupling of a crystal defect to the surrounding vibrational modes results in an effectively classical Fokker–Planck equation (2), with all quantum mechanical effects contained in the
. Equations (2) and (3) show that, in the approximation of a harmonic potential well, the coupling of a crystal defect to the surrounding vibrational modes results in an effectively classical Fokker–Planck equation (2), with all quantum mechanical effects contained in the  dependent effective system temperature
dependent effective system temperature  . Although no exact result exists for the general nonlinear migration potential in the open quantum systems theory setting, an approximate diffusivity DOQS that accounts for quantum zero-point motion can be produced by using the effective temperature
. Although no exact result exists for the general nonlinear migration potential in the open quantum systems theory setting, an approximate diffusivity DOQS that accounts for quantum zero-point motion can be produced by using the effective temperature  in the classical expression for one dimensional motion in a periodic potential [39, 40]
in the classical expression for one dimensional motion in a periodic potential [39, 40]
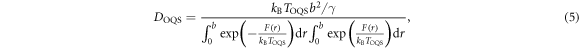
where  is the free energy of migration, given in the harmonic approximation by
is the free energy of migration, given in the harmonic approximation by
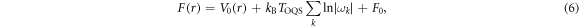
and where F0 is a constant term that does not affect our results. When  is much smaller than the free energy barrier
is much smaller than the free energy barrier  ,
,  is given by the well known expression from the Kramers transition state theory [39]
is given by the well known expression from the Kramers transition state theory [39]
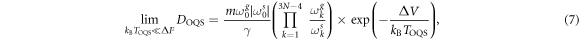
where  and
and  are the frequencies at the ground and saddle point of the static migration potential
are the frequencies at the ground and saddle point of the static migration potential  , taken here either from EAM or DFT calculations, whilst the
, taken here either from EAM or DFT calculations, whilst the  are the eigenfrequencies at the ground state and saddle state [39], which here we take only from EAM calculations. At effective temperatures such that
are the eigenfrequencies at the ground state and saddle state [39], which here we take only from EAM calculations. At effective temperatures such that  we obtain the free diffusion coefficient
we obtain the free diffusion coefficient
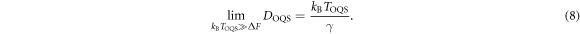
Equation (5) constitutes our main theoretical result, an expression for the migration rate of a crowdion in a heavy atom system, valid down to temperatures of at least 1 K. In figure 2 equation (5) is illustrated using the migration potential  found in molecular statics and DFT calculations. We see that using the migration barrier of 17.2 meV derived from the empirical potential,
found in molecular statics and DFT calculations. We see that using the migration barrier of 17.2 meV derived from the empirical potential,  is in good agreement with the diffusivity extracted from our quantum heat bath molecular dynamics simulations. When using the much lower migration barrier of 2.07 meV found in our DFT calculations, as such a small migration barrier is easily overcome by all effective classical temperatures, the diffusivity is essentially identical at all system temperatures to the free diffusion limiting form (8).
is in good agreement with the diffusivity extracted from our quantum heat bath molecular dynamics simulations. When using the much lower migration barrier of 2.07 meV found in our DFT calculations, as such a small migration barrier is easily overcome by all effective classical temperatures, the diffusivity is essentially identical at all system temperatures to the free diffusion limiting form (8).
Generalization to larger SIA clusters
Whilst we have developed an accurate model for single SIA crowdions, irradiated materials will in practice have a large distribution of SIA cluster sizes [5]. Migration barriers of larger SIA loops have not yet been calculated in DFT due to computational restrictions on the system size; however, from previous classical heat bath molecular dynamics studies of SIA cluster motion in bcc metals, it is known that for larger SIA clusters the migration barrier is approximately constant [11, 15], with a prefactor that scales as the inverse square root of the cluster size N [15]. In addition, above around 200 K the cluster exhibits free Brownian motion [16], where  , with a friction coefficient γ that also scales as
, with a friction coefficient γ that also scales as  . We have performed classical molecular dynamics simulations of a 7-SIA cluster, using trajectories of up to 94 ns at low temperature to obtain statistically robust measurements of the diffusion. We find a migration barrier of around 70 meV at low temperature and a friction coefficient of 13 meV
. We have performed classical molecular dynamics simulations of a 7-SIA cluster, using trajectories of up to 94 ns at low temperature to obtain statistically robust measurements of the diffusion. We find a migration barrier of around 70 meV at low temperature and a friction coefficient of 13 meV  ps−1 at high temperature. However, as our DFT calculations revealed that the crowdion migration barrier was 12% of the EAM value; accounting for this discrepancy yields an SIA cluster migration barrier of 8 meV. The effects of such a small migration barrier will be overcome at effective classical temperatures of
ps−1 at high temperature. However, as our DFT calculations revealed that the crowdion migration barrier was 12% of the EAM value; accounting for this discrepancy yields an SIA cluster migration barrier of 8 meV. The effects of such a small migration barrier will be overcome at effective classical temperatures of  , allowing us to use the high temperature approximation (8) for the loop mobility. Collating the information from ab initio calculations and quantum heat bath molecular dynamics simulations thus gives our main result, the diffusivity of an N-SIA cluster in tungsten
, allowing us to use the high temperature approximation (8) for the loop mobility. Collating the information from ab initio calculations and quantum heat bath molecular dynamics simulations thus gives our main result, the diffusivity of an N-SIA cluster in tungsten
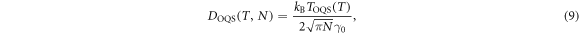
where  meV
meV  ps−1 and the effective temperature
ps−1 and the effective temperature  is formally given by equation (3). A simple form of (9), appropriate for implementation in numerical simulations, can be obtained after two simplifying approximations. Firstly, as
is formally given by equation (3). A simple form of (9), appropriate for implementation in numerical simulations, can be obtained after two simplifying approximations. Firstly, as  is proportional to the magnitude of the Hessian matrix of second derivatives (see supplementary material and [8]) we conclude that
is proportional to the magnitude of the Hessian matrix of second derivatives (see supplementary material and [8]) we conclude that  is proportional to μ, the shear modulus. In addition, we have found that
is proportional to μ, the shear modulus. In addition, we have found that  can be well approximated by the simple function
can be well approximated by the simple function  , where a is a fitting parameter. Under these approximations we obtain the readily usable form
, where a is a fitting parameter. Under these approximations we obtain the readily usable form
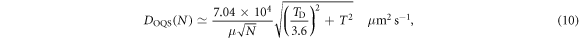
where  GPa and
GPa and  are the shear modulus and Debye temperature of tungsten, giving a numerical value of
are the shear modulus and Debye temperature of tungsten, giving a numerical value of  . We note that the approximation in equation (10) predicts a breakdown in classical behaviour once
. We note that the approximation in equation (10) predicts a breakdown in classical behaviour once  , in agreement with a high temperature expansion of the Bose–Einstein distribution [41].
, in agreement with a high temperature expansion of the Bose–Einstein distribution [41].
Discussion
Our main result, equation (9), provides a predictive calculation of the diffusivity of self-interstitial defects in pure tungsten, valid down to cryogenic temperature, which are presently used in a wide variety of experiments investigating post irradiation microstructure evolution. Due to the influence of zero-point motion, we find that the diffusivity remains very high, of order  for a 100-SIA cluster. This high value, which has never been measured or estimated previously, has clear implications for transmission microscopy experiments conducted in
for a 100-SIA cluster. This high value, which has never been measured or estimated previously, has clear implications for transmission microscopy experiments conducted in  in thick foils at low temperatures [4, 6]. Using a diffusivity of
in thick foils at low temperatures [4, 6]. Using a diffusivity of  we expect clusters to diffuse across typical foil thicknesses in around 0.03 s, leading to a likely depreciation in the cluster population even at cryogenic temperatures in the absence of impurities or other imperfections. Furthermore, as the diffusivity remains constant below 50 K, the effective activation energy is zero and therefore it is unlikely that self-interstitial defect migration will feature in the measured spectrum of energies derived from resistivity recovery experiments.
we expect clusters to diffuse across typical foil thicknesses in around 0.03 s, leading to a likely depreciation in the cluster population even at cryogenic temperatures in the absence of impurities or other imperfections. Furthermore, as the diffusivity remains constant below 50 K, the effective activation energy is zero and therefore it is unlikely that self-interstitial defect migration will feature in the measured spectrum of energies derived from resistivity recovery experiments.
Acknowledgments
We gratefully acknowledge many stimulating discussions with K. Arakawa and M.-C. Marinica concerning the nature of low temperature defect diffusion. This work was stimulated by the preliminary experimental electron microscope observations of low temperature migration of defects reported in [6] and elsewhere. While our work involves a number of approximations, particularly about the absence of impurities or other defects, which in any experimental observation likely have a profound effect on the measured transport properties, we believe that the general approach described above will help with the development of a comprehensive theoretical framework within which experimental data can be interpreted. Density functional calculations were carried out using the EUROfusion Marconi supercomputer facility in Italy. This work has been carried out within the framework of the EUROfusion Consortium and has received funding from the Euratom research and training programme 2014–2018 under grant agreement No 633053 and from the RCUK Energy Programme [grant number EP/P012450/1]. To obtain further information on the data and models underlying this paper please contact PublicationsManager@ukaea.uk. The views and opinions expressed herein do not necessarily reflect those of the European Commission.