

Abstract
Identification and synthesis of 2D topological insulators is particularly elusive. According to previous ab initio predictions 2D InBi (Indium Bismide) is a material exhibiting topological properties which are combined with a band gap suitable for practical applications. We employ ab initio molecular dynamics (AIMD) simulations to assess the thermal stability as well as the mechanical properties such as elastic modulus and stress–strain curves of 2D InBi. The obtained new knowledge adds further characteristics appealing to the feasibility of its synthesis and its potential applications. We find that pristine 2D InBi, H-InBi (hydrogenated 2D InBi) as well as 2D InBi heterostructures with graphene are all stable well above room temperature, being the calculated thermal stability for pristine 2D InBi 850 K and for H-InBi in the range above 500 K. The heterostructures of 2D InBi with graphene exhibit thermal stability exceeding 1000 K. In terms of mechanical properties, pristine 2D InBi exhibits similarities with another 2D material, stanene. The fracture stress for 2D InBi is estimated to be ∼3.3 GPa (∼3.6 GPa for stanene) while elastic modulus of 2D InBi reads ∼34 GPa (to compare with ∼23 GPa for stanene). Overall, the thermal stability, elastic, and fracture resistant properties of 2D InBi and its heterostructures with graphene appear as high enough to motivate future attempts directed to its synthesis and characterization.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Advancement of emergent spintronics involves identification and development of 2D material systems which can host the realization of topological insulators (TI), and a research focus has been placed on material compositions containing heavy elements such as bismuth (Bi) [1, 2]. A family of 2D TIs in binary compositions of group III elements (B, Al, Ga, In, and Tl) and Bi in a buckled honeycomb structure, both pristine and hydrogenated, has been predicted by first-principles electronic structure calculations [3, 4]. Particularly, the buckled honeycomb structure of GaBi, InBi, and TlBi have been found to exhibit a nontrivial band topology characterized by Z2 topological invariants, resulting from strong spin–orbit coupling (SOC) in such systems.
We refer to this family of materials as 2D group III bismides, and by employing ab initio calculations, we have investigated different aspects of their electronic and topologic properties, as well as tuning by chemical functionalization, composition alterations, and strain [5–7]. We have demonstrated that hydrogenation of 2D InBi not only preserves and assists its topological insulating properties but also opens a band gap exceeding 0.32 eV [5]. Such band gap values compare favorably with the thermal energy at room temperature which suggests that 2D H-InBi as well as any other chemically or structurally tuned variants of 2D InBi may constitute a 2D TI with viable prospects for room-temperature applications and device concepts. Choosing different geometric configurations for functionalization (e.g. boat-like, chair-like) as well as electing to apply strain to pristine and chemically functionalized 2D InBi may further favorably modify its electronic and topological properties [5, 7].
For a given material, the feasibility of some of its electronic properties for room temperature applications, e.g. large enough band gap, is unrelated to its thermal stability. However, the thermal stability decisively impacts the feasibility of the material's future applications. Reliable and scalable synthesis, perseverance, manipulation, robustness when submitted to electronic technology engineering operations, and longevity may all depend on thermal stability. Such reasoning makes investigating the thermal stability of potential 2D TIs a well-motivated task. Similar considerations apply to assessing the mechanical properties of 2D InBi and its derivatives whereby any new knowledge about strain–stress relationship and elastic properties (e.g. elastic modulus) is both beneficial for pragmatic purposes of handling potential device development, and useful for assisting efficient synthesis efforts that may necessitate to choose out of a variety of substrates or confinement contexts, as previously succeeded for other pioneering 2D materials as 2D group III nitrides [8–10] and 2D InO [11, 12].
The thermal and mechanical properties of better studied 2D materials have been theoretically approached by huge variety of methods, from phenomenological descriptions based on experimental results to various classical and ab initio solutions, from accounting for substrates and for number-of-layer accumulation to structural evolution during growth and deposition [13].
Ab initio molecular dynamics (AIMD), which due to generally increased availability of computational resources nowadays becomes computationally more affordable, represents an excellent compromise between accuracy and efficiency for modeling the thermal and mechanical properties of newly envisaged 2D materials. This is especially so when concerning novel 2D materials that are yet to be synthesized and for which there are no experimental results to serve for any model parametrization.
2D compounds consisting in atoms of heavy elements such as In and Bi are not well studied and understood yet. This is especially so regarding their thermal and mechanical features. In this work, we employ advanced AIMD simulations to estimate the thermal stability as well as the stress–strain relationship and elastic moduli for: (i) pristine 2D InBi; (ii) a functionalized variant of 2D InBi, namely different isomers of hydrogenated 2D InBi (H-InBi); and (iii) two heterostructures containing 2D InBi: a 2D InBi on top of a graphene sheet, and a 2D InBi confined between two graphene sheets, this last heterostructure representing a simplified model of 2D InBi in confinement.
We find that pristine 2D InBi and the different isomers of H-InBi are all stable well above room temperature with the heterostructures of InBi exhibiting high thermal limit exceeding 1000 K which may assist choosing appropriate substrates for deposition of 2D InBi as well as incentivize its synthesis attempts in confinement. In terms of mechanical properties, pristine 2D InBi records a fracture stress of ∼3.3 GPa in both in-plane (x and y) directions and elastic modulus value of 34 GPa in both in-plane (x and y) directions. Such values are comparable to those calculated by others for another novel 2D semiconductor material which is also claimed to exhibit TI properties—the 2D stanene [14] which is characterized by fracture stress 3.6 GPa and elastic modulus 23 GPa, respectively [15]. Our results indicate that although less mechanically robust than, e.g. 2D h-BN (hexagonal BN) [15] and graphene [16, 17], which are currently perceived as the two mechanically most robust 2D materials, the 2D InBi exhibits elastic and fracture resistant properties comparable to other emerging 2D materials and perceived as high enough to motivate pursuing its synthesis, characterization, and manipulation attempts.
2. Methodology and computational details
Thermal stability and mechanical properties of nanostructured materials are usually investigated by molecular dynamics simulations. Classical molecular dynamics is a computationally efficient method to approach such simulations. Full periodic table force field parametrizations for molecular dynamics simulations, based e.g. on the Lennard-Jones potential [18], have existed for a long time and are quite successful when describing 2D materials which composition involves lighter and better studied elements such as 2D SiC [19]. However, when the 2D material is composed by heavier elements such as In and Bi with corresponding parametrizations that are less or not at all tested in molecular dynamics simulations of 2D structures, then such parametrizations are not able to reproduce even basic equilibrium structures and bonding features, as our preliminary tests based on [18] have demonstrated. Another potential, widely used for molecular dynamics simulations of 2D materials is the many-body Tersoff potential [20, 21]. Again, no widely tested Tersoff potentials for the elemental pair of In and Bi are presently available.
At the same time, AIMD has become a state-of-the-art method for addressing thermal and mechanical properties of 2D materials. In this work we carry out AIMD simulations employing the VASP code [22] using the projector augmented wave (PAW) [23] method. Van der Waals (vdW) corrections [24, 25] have been employed for achieving higher accuracy and a deeper understanding at atomistic level of processes such as thermal disintegration, delamination, etc. Naturally, vdW corrections have been taken into account only in the AIMD simulation of heterostructures considered in this work.
The initial structural configurations for pristine 2D InBi and H-InBi that we employ in the AIMD simulations were obtained in our previous modelling of the 2D InBi material system [5, 6]. These structures have been relaxed using Perdew−Burke−Ernzerhof (PBE) [26] generalized gradient approximation level of theory, and PAW method, as implemented in VASP, and the structure optimization details as well as the motivation for present choice of level of theory are discussed in [5–7]. While it is essential that SOC is explicitly taken into account in the band structure calculations and for reliably assessing TI properties of compounds containing a heavy element such as Bi [5–7], for the purposes of AIMD calculations dedicated to their thermal stability and mechanical properties, SOC inclusion is not perceived as a necessary requirement.
Concerning the VASP/PBE calculations as performed at each MD step and in order to avoid spurious interactions with images of the supercells of the model systems representing 2D InBi, its hydrogenated counterparts, and the investigated heterostructures, a sufficiently large vacuum space (25 Å) above and below these 2D model systems was ensured.
The gestation of the AIMD simulations, as well the corresponding data post-processing and its visualization were carried out within the Atomic Simulation Environment [27].
As usual for the purposes of thermal and structural properties, for all AIMD results reported here, to control the temperature of the model systems we use the NVT environment through the Nosé–Hoover thermostat. Length of time steps varies and is adapted to the specificity of the concrete simulation, from 1 fs adopted for the more complex model systems, such as H-InBi as well as the heterostructures involving InBi, up to 5 fs as a compromise for performing test calculation as well as longer simulation runs for the pristine InBi. In stress–strain calculations, the stress tensor was calculated for three degrees of freedom of the ionic positions. For simulations including vdW corrections, the optB88-vdW option was used [24, 25].
To ensure that the results obtained do not depend on the choice of supercell size, extensive tests with different supercell sizes matched with different sets of k-point meshes have been conducted. As a result of this testing, we found that the use of relatively small supercell size together with denser k-point meshes provides similar or better accuracy to the accuracy provided by simulations employing larger supercells and single k-point. All results reported here are thus obtained by employing relatively small supercell sizes with denser k-point meshes that have been previously optimized for the 2D InBi system and its derivates at the same level of DFT as the one used in this work [5–7].
Two reasons motivate why graphene is also submitted to the AIMD simulations carried out here. Induced strain in pristine single graphene sheet was calculated in order to be compared to the vast amount of data available in the literature on this subject [16, 17, 20] for test purposes, i.e. to verify to what extent the adopted here AIMD simulation environment, DFT theory levels for the force field, as well as simulation parameters selected, perform well in such calculations involving 2D material systems. Another obvious reason to submit graphene to AIMD simulations of induced strain is that it constitutes an essential part of the InBi heterostructures investigated in this work.
Stress calculations were carried out firstly without any induced strain so to acquire how much stress is present in the 2D model system due to its thermal vibrations. This inherent thermal stress is then subtracted from the total stress to only evaluate the stress from the induced strain. Technically, stress is calculated at each AIMD simulation step. The stress is then summed up and averaged for the whole length of the simulation after reaching equilibrium. As usual, the elastic modulus is calculated from the slope of the stress–strain curve for low values of induced strain where the stress–strain relation is presumably linear.
3. Results and discussion
3.1. Thermal stability and mechanical properties of pristine 2D InBi
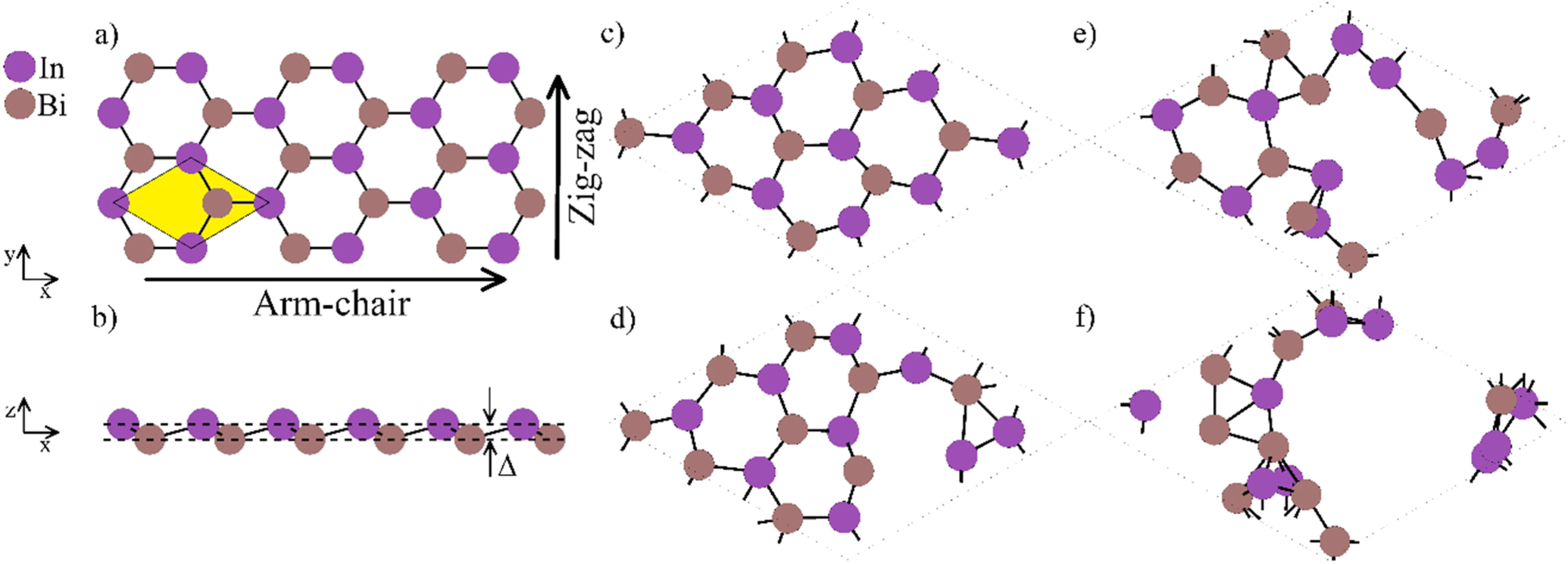
A comprehensive analysis of ab initio simulation results on the structure of pristine 2D InBi reveals it as a hexagonal low-buckled sheet with lattice parameter a = 4.922 Å, a buckling parameter of 0.85 Å, and In–Bi bond length of 2.844 Å (figures 1(a), (b)). The pristine 2D InBi exhibits an indirect band gap of 0.175 eV with band inversion characterizing it as a 2D topological insulator [5, 6].

Figure 1. (a) Top view of pristine 2D InBi sheet. The area marked in yellow represents the unit cell. Arm-chair and zig-zag directions of the honeycomb 2D structure are also indicated; (b) Side view of the of pristine 2D InBi sheet with the buckling parameter Δ indicated; (c)–(f) Structural snapshots of pristine 2D InBi disintegrating at 850 K (2000 time steps of 5 fs each): (c) step 28; (d) step 233; (e) step 675; and, (f) step 1028.
Download figure:
Standard image High-resolution imageThe AIMD calculation of the thermal stability of pristine 2D InBi employed supercells (3 × 3 × 1). Simulations ran for 2000 time steps (time step length of 5 fs). Large sets of test calculations were caried out for the interval of temperatures between 1 and 1000 K and then in gradually decreasing temperature intervals in order to determine with precision of 2 K the temperature at which 2D InBi thermally disintegrates. We found that it remains stable for temperatures lower or equal to 848 K (including at AIMD test runs employing different time scales and different time step lengths). At the temperature of 850 K, 2D InBi gradually but quickly disintegrates as shown in figures 1(c)–(f). The first broken and rearranged bonds in the pristine 2D InBi sheet are observed at around step 233 or about ∼1200 fs = 1.2 ns after the start of the simulation, figure 1(d). Thermal disintegration of the 2D InBi progresses then at increased speed, with only few hexagons of its honeycomb sheet surviving as intact structures at step 675, figure 1(e), and by step 1028, the 2D InBi sheet is nearly fully disintegrated. This result, obtained by AIMD and indicating that pristine 2D InBi remains thermally stable up to 850 K (577 °C), is certainly promising for any future synthesis and characterization efforts to be dedicated to 2D InBi.
By carrying out sets of AIMD simulations runs (after tests setting again to 2000 timesteps with duration of 5 fs), we have studied induced strain in 2D InBi. The induced strain was simulated at 5 different temperatures: 1 K, 100 K, 300 K, 500 K, and 800 K. Mechanical properties such as the elastic modulus, the fracture stress, and the fracture strain are then calculated from the strain–stress curves. As it is well-known from comprehensive modelling of other 2D materials with honeycomb structure, their mechanical properties may be significantly different in arm-chair and zig-zag direction, respectively. This is particularly true for some of the binary 2D materials such as SiC [15]. Consequently, we calculated the stress corresponding to a given strain, i.e. the strain–stress relation, and we obtain the respective strain–stress curves for pristine 2D InBi in both its arm-chair (figure 2(a)) and its zig-zag (figure 2(b)) direction and at the temperatures of 1 K, 100 K, 300 K, 500 K, and 800 K.

Figure 2. Induced strain of pristine 2D InBi at 1 K, 100 K, 300 K, 500 K, and 800 K in: (a) arm-chair; and, (b) zig-zag direction.
Download figure:
Standard image High-resolution imageFrom the strain–stress curves (figures 2(a) and (b)), the elastic modulus, the fracture stress, and the fracture strain (ε)/ / for the pristine 2D InBi in its arm-chair and its zig-zag directions and for the corresponding temperatures are obtained (table 1). It is noteworthy, that the elastic modulus, the fracture stress and the fracture strain of pristine 2D InBi remain practically the same independently of the direction in which they are simulated (arm-chair or zig-zag) up to room temperature and above (300 K). First at temperature of 800 K, which is close to the temperature of thermal disintegration of the material (850 K), noticeable differences in the elastic modulus and the fracture strain of about 11% and about 5% between the zig-zag and the arm-chair directions emerge. At temperatures about room temperatures and up to 500 K the zig-zag direction of 2D InBi is only very marginally more fracture resistant than its arm-chair direction (cf, table 1).
/ for the pristine 2D InBi in its arm-chair and its zig-zag directions and for the corresponding temperatures are obtained (table 1). It is noteworthy, that the elastic modulus, the fracture stress and the fracture strain of pristine 2D InBi remain practically the same independently of the direction in which they are simulated (arm-chair or zig-zag) up to room temperature and above (300 K). First at temperature of 800 K, which is close to the temperature of thermal disintegration of the material (850 K), noticeable differences in the elastic modulus and the fracture strain of about 11% and about 5% between the zig-zag and the arm-chair directions emerge. At temperatures about room temperatures and up to 500 K the zig-zag direction of 2D InBi is only very marginally more fracture resistant than its arm-chair direction (cf, table 1).
Table 1. Elastic modulus (GPa), fracture stress (GPa), and the fracture strain (ε) at 1 K, 100 K, 300 K, 500 K, and 800 K of the pristine 2D InBi in its arm-chair and its zig-zag directions.
| Temperature quantity | 1 K | 100 K | 300 K | 500 K | 800 K |
|---|---|---|---|---|---|
| Arm-chair direction of 2D InBi | |||||
| Elastic modulus | 36.851 | 34.465 | 34.132 | 30.758 | 24.733 |
| Fracture stress | 4.305 | 3.868 | 3.273 | 2.373 | 0.436 |
| Fracture strain | 0.196 | 0.153 | 0.124 | 0.102 | 0.022 |
| Zig-zag direction of 2D InBi | |||||
| Elastic modulus | 36.406 | 34.466 | 34.250 | 30.258 | 22.243 |
| Fracture stress | 4.725 | 4.086 | 3.345 | 2.428 | 0.414 |
| Fracture strain | 0.240 | 0.176 | 0.130 | 0.102 | 0.022 |
For comparative purposes, in table 2, the elastic moduli, fracture stresses and fracture strain values as obtained in this work for the pristine 2D InBi and for graphene (both at 300 K) are listed together with the same quantities obtained in [15] by employing classical molecular dynamics simulations making use of Lennard-Jones potentials and for the temperature of 250 K for 2D BN, 2D SiC, and 2D stanene. While 2D boron nitride (BN) is the most fracture resistant 2D material, graphene exhibits the highest elastic modulus, and 2D SiC is another stiff and strong 2D material, other 2D materials as e.g. stanene are less stiff (lower elastic modulus) and less strong (lower fracture stress). Still, not only stanene has been successfully synthesized in supported heterostructures and even as a free-standing sheets [28, 29] but it is perceived as a prospective electronic and topological insulator 2D material [30]. Our results reveal the pristine 2D InBi as a 2D material slightly less fracture resistant and about 40% stiffer than stanene (cf, table 2, n.b., 2D InBi properties were simulated at 300 K while stanene properties were simulated at 250 K [15]).
Table 2. Elastic modulus (GPa), fracture stress (GPa) and the fracture strain (ε) as obtained in this work for the pristine 2D InBi and for graphene (both at 300 K) compared to the same quantities obtained in [15] (at 250 K) for 2D BN, 2D SiC, and 2D stanene.
| Quantity 2D material | Direction | Elastic modulus | Fracture stress | Fracture strain |
|---|---|---|---|---|
| Graphene (this work) | arm-chair | 913.40 | 99.50 | 0.198 |
| zig-zag | 898.52 | 110.06 | 0.242 | |
| BN [15] | arm-chair | 715 | 106.4 | 0.262 |
| zig-zag | 727 | 112.48 | 0.291 | |
| SiC [15] | arm-chair | 650 | 75.5 | 0.124 |
| zig-zag | 510 | 56 | 0.126 | |
| Stanene [15] | arm-chair | 23 | 3.57 | 0.179 |
| zig-zag | 24 | 3.6 | 0.207 | |
| InBi | arm-chair | 34.132 | 3.273 | 0.124 |
| zig-zag | 34.25 | 3.345 | 0.130 |
3.2. Thermal stability and mechanical properties of 2D H-InBi
As previously discussed in detail, the hydrogenation of pristine 2D InBi leads to further enlarging of its band gap from ∼0.175 up to ∼0.271–0.320 eV (depending on the configuration of the hydrogenated structures) thus placing the hydrogenated 2D InBi (H-InBi) firmly in the category of room temperatures 2D semiconductors [5]. This justifies investigating whether and how the hydrogenation of 2D InBi affects its thermal stability. We investigate two configurations of the 2D H-InBi, chair-like and boat-like, as shown in figure 3.

Figure 3. (a) Top view of H-InBi in the chair-like configuration. The area marked in yellow represents the unit cell. Arm-chair and zig-zag directions of the structure are also indicated; (b) side view of the of H-InBi in the chair-like configuration; (c) top view of H-InBi in the boat-like configuration; (d) side view of the of H-InBi in the boat-like configuration.
Download figure:
Standard image High-resolution imageThe chair-like configuration of 2D H-InBi (figures 3(a) and (b)) is similar to the pristine 2D InBi (figures 1(a), (b)), the difference residing in the hydrogen bonds distributed for In and Bi atoms on the opposite sides of the 2D sheet. The side view of the boat-like configuration of 2D H-InBi (figure 3(d)) differs in conformation from the pristine 2D InBi and the chair-like 2D H-InBi by exhibiting different orientations of the hydrogen bonds due to both In and Bi atoms exhibiting hydrogen bonds on the same side of the sheet, but switching side at the arm-chair profile of the sheet. Buckling parameter for the 2D H-InBi increases from 0.85 (pristine 2D InBi) to 0.902 Å.
The AIMD simulations of the thermal stability of 2D H-InBi follow a similar calculation scheme as applied to pristine 2D InBi (section 3.1).
The AIMD simulations indicate that the chair-like H-InBi thermally disintegrates at 664 K while the boat-like H-InBi thermally disintegrates at 500 K. Thus, for both the chair-like H-InBi and the boat-like H-InBi configurations the thermal stability decreases in comparison to the pristine 2D InBi.
It may look contra-intuitive that passivation of the dangling bonds of certain structure lowers its thermal stability, however in the case of hydrogenation of 2D InBi (as it also happens for other 2D binary materials) also the bond environment and structure conformation change, which may explain the slightly lower thermal stability of the 2D H-InBi. In any experimental context of synthesis and characterization of 2D InBi, where any partial hydrogenation or, for that matter, any other type of functionalization may be present, these results should be perceived qualitatively. Namely, hydrogenation may somewhat lower the thermal stability of the 2D InBi but the temperatures at which its thermal disintegration is expected to happen, still remain with at least 200 K (and more) above the room temperature.
Following the same procedure as for pristine 2D InBi, we apply AIMD to address the mechanical properties of 2D H-InBi in its chair-like and boat-like configurations and in both their arm-chair and zig-zag directions. Considering that the thermal dependence of the mechanical properties of pristine 2D InBi (figure 2 and table 1) is quite insignificant (except at temperatures close to thermal disintegration), and not unexpected, the induced strain for 2D H-InBi was simulated at two different temperatures 1 K and 300 K. The values of the elastic modulus, the fracture stress, and the fracture strain for 2D H-InBi in its chair-like and boat-like configurations and in both their arm-chair and zig-zag directions are then calculated from the strain–stress curves (figure 4) and listed in table 3.

Figure 4. Induced strain of 2D H-InBi at 1 K, and 300 K in: (a) 2D H-InBi chair-like configuration in arm-chair direction; (b) 2D H-InBi chair-like configuration in zig-zag direction; (c) 2D H-InBi boat-like configuration in in arm-chair direction; (d) 2D H-InBi boat-like configuration in in zig-zag direction.
Download figure:
Standard image High-resolution imageTable 3. Elastic modulus (GPa), fracture stress (GPa), and the fracture strain (ε) at 1 K and 300 K, of the 2D H-InBi in its chair-like and boat-like configurations and in both their arm-chair and zig-zag directions.
| Temperature quantity | 1 K | 300 K |
|---|---|---|
| Arm-chair direction of 2D H-InBi boat-like configuration | ||
| Elastic modulus | 15.716 | 11.153 |
| Fracture stress | 2.04 | 1.26 |
| Fracture strain | 0.182 | 0.120 |
| Zig-zag direction of 2D H-InBi boat-like configuration | ||
| Elastic modulus | 26.605 | 23.184 |
| Fracture stress | 3.33 | 2.20 |
| Fracture strain | 0.196 | 0.142 |
| Arm-chair direction of 2D H-InBi chair-like configuration | ||
| Elastic modulus | 34.872 | 27.461 |
| Fracture stress | 3.58 | 2.25 |
| Fracture strain | 0.156 | 0.108 |
| Zig-zag direction of 2D H-InBi chair-like configuration | ||
| Elastic modulus | 33.544 | 26.131 |
| Fracture stress | 3.99 | 2.48 |
| Fracture strain | 0.178 | 0.114 |
The chemical nature of 2D H-InBi significantly decreases the elastic modulus, fracture stress and fracture strain in both its chair-like and boat-like configurations compared to pristine 2D InBi. Comparing the H-InBi in both configurations, it is found that the chair-like H-InBi fractures at higher stress while the boat-like H-InBi fractures at higher strain. The elastic modulus of 2D H-InBi is higher in its chair-like configuration. This shows that its chair-like configuration is stiffer and stronger than the more elastic boat-like configuration. Looking specifically at the boat-like 2D H-InBi configuration, it is stronger and more elastic in its zig-zag direction than in its arm-chair direction, while when looking specifically at the chair-like 2D H-InBi configuration, its zig-zag and arm-chair direction are comparably strong and elastic. These differences deduced from the present simulations may be useful in case of any future synthesis attempts favoring a given configuration (e.g. the chair-like) in contrast to its alternative.
3.3. Thermal stability and mechanical properties of heterostructures with 2D InBi
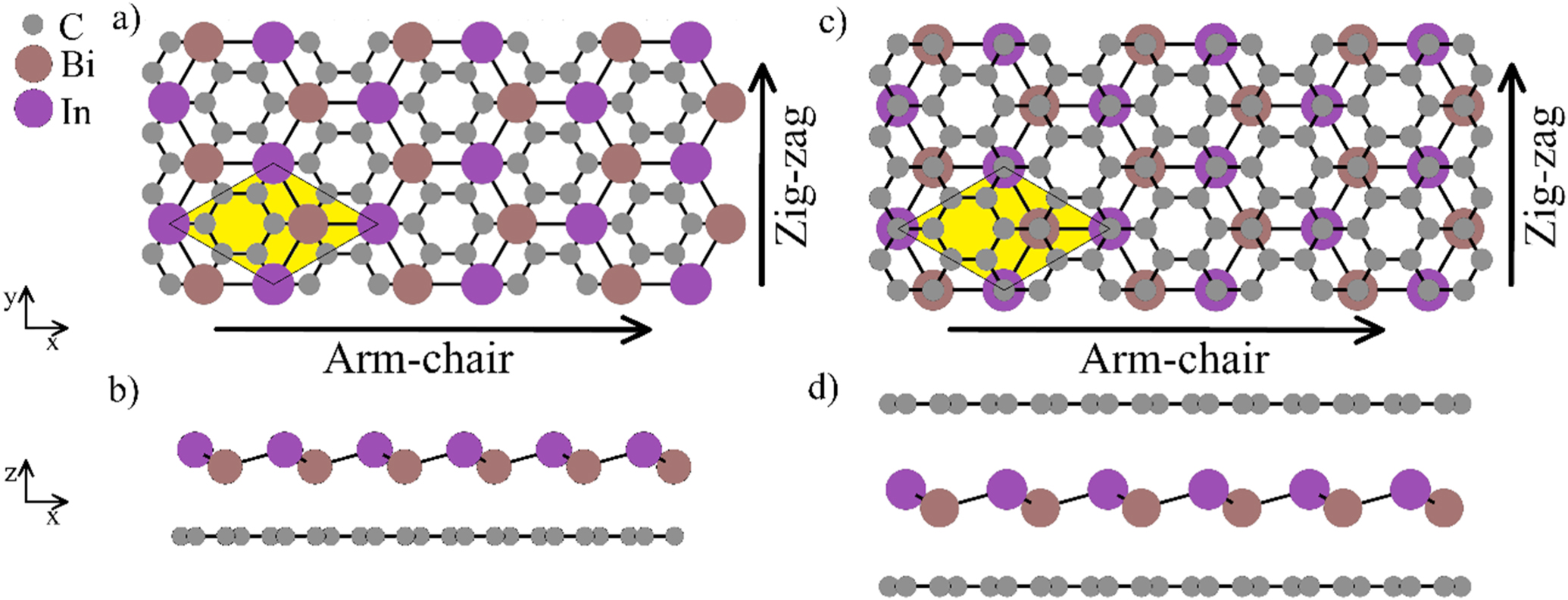
To approximate any hypothetical experimental situations of synthesizing 2D InBi, a heterostructure consisting in a 2D InBi on top of graphene sheet was studied by AIMD (figures 5(a) and (b)). In addition, and to approximate the synthesis of 2D InBi in confinement by pursuing the approach that succeeded to achieve 2D group III nitrides [8–10] and 2D InO [11, 12], the heterostructure consisting in a 2D InBi sheet in confinement between two graphene sheets was also simulated (figures 5(c) and (d)).

Figure 5. Heterostructures of 2D InBi with graphene: (a) top view of a 2D InBi on top of graphene sheet. Arrows indicate the arm-chair and zig-zag directions. The yellow area represents the unit cell; (b) side view of the of a 2D InBi on top of graphene sheet; (c) top view of 2D InBi sheet in confinement between two graphene sheets; (d) side view of the of 2D InBi sheet in confinement between two graphene sheets.
Download figure:
Standard image High-resolution imageThermal stability of both heterostructures of 2D InBi with graphene (figure 5) was studied by running sets of AIMD simulations for 4000–8000 time-steps with time step length of 1 fs. The mechanism of thermal disintegration is shared for both the 2D InBi on top of graphene sheet (figures 5(a) and (b)) and the 2D InBi sheet in confinement between two graphene sheets (figures 5(c) and (d)) and consists in delamination of the graphene sheet(s) from the 2D InBi sheet. Delaminated graphene sheets preserve their structural integrity during and after the delamination. This heterostructure delamination and subsequent 2D InBi disintegration occurs at 1702 K for the 2D InBi on top of graphene sheet, and at 1232 K for the 2D InBi sheet in confinement between two graphene sheets.
Increased thermal stability of 2D materials when incorporated in vdW heterostructures has been reported previously, as in the case of phosphorene in confinement between two graphene sheets studied by molecular dynamics [31].
The result that the heterostructure consisting in 2D InBi on top of a graphene sheet exhibits a higher thermal stability than the heterostructure consisting in a 2D InBi confined between two graphene sheets may look counterintuitive. One tentative explanation may reside in the fact that in both cases the thermal disintegration starts as delamination. Once delamination starts happening, the disintegration of the 2D InBi is almost instantaneous due to temperatures, in both cases of these heterostructures, much higher (above 1000 K) than the thermal stability of a free-standing pristine 2D InBi (850 K). In comparison to the heterostructure consisting in an InBi layer on top of graphene, the confined heterostructure relies (for holding together) on vdW forces on both sides of the 2D InBi sheet which could make it prone to delamination at lower temperature.
The increased thermal stability of the heterostructures of 2D InBi with graphene is due (i) to efficient dissipation of the thermal energy in the graphene sheet(s) due to its high thermal conductivity [32], and (ii) to the high thermal stability of graphene itself.
The results for thermal stability of heterostructures of 2D InBi with graphene should be perceived as upper limits for thermal stability of such 2D architectures held together by the vdW forces. Real experimental situations of heterostructures with 2D InBi would certainly contain defects, vacancies, etc, which lower the thermal stability. In addition, the experimental approach to synthesis of 2D InBi in confinement may rely on different heterostructures, for example, like the ones employed for 2D group III nitrides [8–10] and 2D InO [11, 12] where the 2D material is confined between SiC substrate and a graphene capping layer. Thus, the essentialconclusion of the presented here AIMD results is that heterostructures with 2D InBi exhibit an increased thermal stability compared to pristine and hydrogenated 2D InBi, possibly above 1000 K.
Out of the different heterostructures involving a 2D InBi sheet we choose to investigate as a case study and because of its foreseen relevance to experimental situations [8–12], the mechanical properties of the heterostructure consisting in 2D InBi sheet in confinement between two graphene sheets, figure 5(b). The induced strain for this heterostructure was simulated at two different temperatures 1 K and 300 K and in its arm-chair and zig-zag directions resulting in the respective strain–stress curves (figure 6). The corresponding values of the elastic modulus, the fracture stress, and the fracture strain are then calculated from the strain–stress curves in figure 6, and listed in table 4.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. Induced strain of 2D InBi sheet in confinement between two graphene sheets at 1 K, and 300 K in: (a) arm-chair direction; (b) zig-zag direction.
Download figure:
Standard image High-resolution image{kind=link}
Table 4. Elastic modulus (GPa), fracture stress (GPa), and the fracture strain (ε) at 1 K and 300 K of 2D InBi sheet in confinement between two graphene sheets at 1 K, and 300 K in in their arm-chair and zig-zag directions.
| Temperature quantity | 1 K | 300 K |
|---|---|---|
| Arm-chair direction of 2D InBi sheet in confinement between two graphene sheets | ||
| Elastic modulus | 592.71 | 566.27 |
| Fracture stress | 62.44 | 60.13 |
| Fracture strain | 0.21 | 0.21 |
| Zig-zag direction of 2D InBi sheet in confinement between two graphene sheets | ||
| Elastic modulus | 552.63 | 551.15 |
| Fracture stress | 69.25 | 67.15 |
| Fracture strain | 0.26 | 0.25 |
At 300 K, the elastic modulus, fracture stress, and fracture strain of 2D InBi in confinement between two graphene sheets (table 4) exhibits markedly higher values (551.15 GPa, 67.15 GPa, 0.25) than the corresponding values of 34.250 GPa, 3.345 GPa, 0.130 for pristine 2D InBi (table 1), and 23.184 GPa, 2.2 GPa, 0.142 (boat-like configuration) and 26.131 GPa, 2.48 GPa, 0.114 (chair-like configuration) for 2D H-InBi (table 3). Thus, the mechanical properties of a confined 2D InBi sheet seem to strongly benefit from a confined configuration and because of the interplay between the confined and confining components of such heterostructure. Yet, the confined heterostructure exhibits inferior mechanical properties when compared to a single graphene sheet itself (cf, tables 1 and 4): the weak bonding by vdW forces of the 2D InBi sheet to graphene may cause the heterostructure to break at lower stress/strain and to exhibit lower elastic modulus than its strongest and most elastic component, the graphene sheet itself. To put these mechanical property results in a wider comparative context, 2D InBi in confinement between two graphene sheets is comparable in elastic modulus and fracture stress, and superior in fracture strain, to 2D SiC (cf, table 1). Moreover, the calculated values of the elastic modulus, fracture stress, and fracture strain of 2D InBi in confinement do not change significantly with the change of the temperature from low values and up to room temperature indicating that the heterostructure of 2D InBi in confinement remains elastic and fracture resistant in a wide temperature range.
4. Conclusions
We employ AIMD simulations to acquire systematic new knowledge on the thermal stability and the mechanical properties of the prospective 2D TI InBi in different configurations: pristine, hydrogenated, and in heterostructures with graphene. Pristine 2D InBi is thermally stable up to 850 K. Its elastic modulus, fracture stress, and fracture strain, in its zig-zag direction and at room temperature read 34.250 GPa, 3.345 GPa, 0.130, which are values not dissimilar to another novel 2D TI, the experimentally confirmed stanene. For hydrogenated 2D H-InBi thermal stability decreases to certain extent, but still remains comfortably above room temperature and in the range above 500 K depending on the configuration of the hydrogenated structures. Hydrogenation also somewhat reduces the mechanical properties of the material which for the zig-zag direction of its chair-like configuration and at room temperature remain the ranges of ∼26 GPa, ∼2.5 GPa, ∼0.11 for the elastic modulus, fracture stress, and fracture strain, respectively. Remarkably, heterostructures of 2D InBi with graphene, and especially those consisting in 2D InBi in confinement emerge as thermally stable in the range of 1000 K and with improved elastic properties and fracture toughness exhibiting values of up to ∼550 GPa, ∼67.15 GPa, ∼0.25 at room temperature for the elastic modulus, fracture stress, and fracture strain, respectively. Experimentally relevant factors unaccounted for in the reported here simulations, such as growth kinetics and nearly unavoidable defect (e.g. vacancies) incorporation during growth would most likely lower both the thermal stability and the mechanical properties of any synthesized structures, but the predicted favorable values leave a wide margin for such variation without affecting their potential to practical applications. Our results also indicate that confinement of 2D InBi between two graphene sheets may result in a heterostructure comparable in elastic modulus and fracture stress to 2D SiC which is considered a strong and elastic material in the realm of 2D materials. In the context of recent successes of synthesis by MOCVD of novel 2D group III nitrides and group III oxides in confinement, the presented AIMD results exacerbate the motivated reasoning behind attempting the synthesis of 2D InBi in confinement by employing MOCVD or any other appropriate synthesis approach.
Acknowledgments
GKG and AKG gratefully acknowledge support by VR2017-04071. The authors also acknowledge resources provided by the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Center (NSC) in Linköping (SNIC 2020/5-146 and SNIC 2020/14-17) partially funded by the Swedish Research Council through Grant agreement No. 2018-05973.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).