

Abstract
Proficient capture of carbon dioxide (CO2) is considered to be a backbone for environment protection through countering the climate change caused by mounting carbon content. Here we present a comprehensive mechanism to design novel functional nanostructures capable of capturing a large amount of CO2 efficiently. By means of van der Waals corrected density functional theory calculations, we have studied the structural, electronic and CO2 storage properties of carbon nitride (g-C6N8) nano sheets functionalized with a range of transition metal (TM) dopants ranging from Sc to Zn. The considered TMs bind strongly to the nano sheets with binding energies exceeding their respective cohesive energies, thus abolishing the possibility of metal cluster formation. Uniformly dispersed TMs change the electronic properties of semiconducting g-C6N8 through the transfer of valence charges from the former to the latter. This leaves all the TM dopants with significant positive charges, which are beneficial for CO2 adsorption. We have found that each TM's dopants anchor a maximum of four CO2 molecules with suitable adsorption energies (−0.15 to −1.0 eV) for ambient condition applications. Thus g-C6N8 nano sheets functionalized with selected TMs could serve as an ideal sorbent for CO2 capture.
Export citation and abstract BibTeX RIS
Introduction
A constant increase in energy demands over the years has accounted for the enhanced consumption of fossil fuels, causing drastic changes in the atmosphere due to the emission of greenhouse gases like CO2. Although there is a strong surge for the use of clean energy, fossil fuels might continue to be the main contributor for at least the near future. Apart from fossil fuels, many industrial processes also contribute a large amount of CO2 to the atmosphere. The capture of atmospheric CO2 is also significant for alleviating the mesosphere as almost half of the emitted CO2 cannot escape and remains in the atmosphere, which is yet another problem to be solved [1]. Thus for the protection of environment and restricting a sharp rise in the global temperature, an efficient capture of CO2 is of immense importance [2, 3]. A modern technology of capturing and storing CO2, called CCS, is believed to be effective in reducing CO2 drastically and it has been implemented recently to a great effect [4]. However for a viable implementation of CCS ,an efficient capture of CO2 is of great significance. Another way to adsorb CO2 has been presented recently by Shi et al through chemical reaction by means of varying the water quantities in nanopores that would promote the hydrolysis process of CO32− [5].
Owing to a possible interaction between CO2 and various carbon-based nanostructures, material-based CO2 storage seems to be a viable option and has been studied extensively in recent times [6–9]. However the choice of a proficient material capable of anchoring multiple CO2 molecules with suitable binding energies is yet another hurdles to cross for efficient CO2 storage. Moreover a deep understanding of the intrinsic mechanism of CO2 interaction with the host material is of great importance and plays an important role in designing an efficient CO2 sorbent [10, 11].
Two-dimensional (2D) nanostructures have lately attracted interest due to their promise in various basic and applied scientific endeavors post the successful synthesis of graphene, a truly single sheet of carbon atoms [12–15]. The maximized surface-to-volume ratio of graphene is particularly appealing to chemical gas sensors to achieve the ultimate level of gas binding. However the constraints on the large-scale production of graphene, and the negligibly weak binding of CO2, compels researchers to go for not only different strategies in enhancing CO2–graphene binding but also explore other post graphene 2D materials.
Tawfik et al recently employed first principles calculations to study the CO2 storage properties of transition metal (TM) doped graphene [16]. They have considered as many as 16 different graphene systems with TMs bonded strongly to defected graphene, capable of anchoring multiple CO2 molecules with reasonable binding energies. Another computational study by Fischer et al explained the interaction of CO2 with TiO2 supported graphene and graphene-oxide sheets by means of density funcional theory (DFT) [17]. It was concluded that the graphene/graphene-oxide supported TiO2 clusters largely influence the binding of CO2. In contrast to TMs and TiO2, Koo et al used calcium (Ca) to functionalize graphene nanoribbons (GNRs) and studied the doped systems as efficient and selective CO2 storage media though first principles calculations based on DFT [18]. Each Ca on GNRs was found to adsorb three CO2 molecules selectively among H2, N2 and CH4 with an average binding energy of 0.80 eV/CO2. Another computational study by Sophia et al investigated the interaction of CO2 with that SrTiO3 (001) surface in connection to the band gap modulation of the latter [19].
The CO2–SrTiO3 (001) binding falls into the chemisorption range with the opening of the band gap of the surface, which differ with the CO2–TiO2 interaction. A relatively different approach was used by Tan et al to capture CO2 by borophene nano sheets with the help of a charge-modulated mechanism [20]. By means of first principles calculations it was found that the borophene nano sheet could anchor multiple CO2 molecules with reasonable binding energy upon getting extra electrons injected into the conductive sheet. Controlled, fast and reversible CO2 adsorption/desorption could be achieved by using negatively charged borophene nano sheets, which makes it one of the excellent materials for CO2 capture. There are few other reports discussing CO2 adsorption by means of MoS2 through electric field control [21] and TM functionalized benzene-complexes [22].
Graphitic carbon nitride (g-C6N8) is an important member of graphene-like 2D materials with uniform pores. It belongs to a family of porous 2D membranes, most of which are experimentally synthesized and have found their applications in various scientific and technological fields [23–28]. Different configurations of carbon nitride have already been investigated as efficient hydrogen storage materials both experimentally and theoretically [29–31]. These studies report that metal-doped carbon nitride sheets turn out to be excellent medium for ambient conditions hydrogen storage. Motivated by this property, we have investigated the promise of g-C6N8 as a high capacity CO2 storage material. Our van der Waals corrected DFT calculations revealed that several TM dopants like Sc to Zn, bind strongly with g-C6N8, and anchor multiple CO2 molecules with a binding energy that is much higher than its value on a pristine sheet.
Computational details
The first principles calculations carried out throughout this project by using VASP code were based on DFT [32–34]. The electronic exchange-correlations interactions were described by means of the generalized gradient approximation (GGA) of Perdew-Burke-Ernzerhof whereas projector augmented-wave pseudopotentials were employed to consider the core electronic effects [35, 36]. Underestimation in the CO2-g-C6N8@TMs bindings, which was caused by the GGA were taken care by including van der Waals correction of Grimme's DFT-D2 as implemented in VASP [37]. An energy cut-off of 500 eV was used for the structural optimization under the plane wave basis set. The Monkhorst-Pack scheme was used for the Brillouin zone sampling with 3 × 3 × 1 k-points for geometry optimization and a 7 × 7 × 1 k-point mesh for plotting the density of states (DOS). Convergence benchmarks of 10−5 eV were used for the relaxation process of the atomic positions and lattice constants, whereas a force criteria of 0.001 eV Å−1 was used. We used a reasonably large 2 × 2 supercell of g-C6N8 with 56 atoms in this study having a vacuum space of 15 Å along the z-direction to avoid the possible vertical interactions. A single dopant of selected TMs was considered on a g-C6N8 supercell that constitutes a relatively small doping concentration of 1.785%.
The binding energies of TMs on g-C6N8 can be calculated by the following relation:
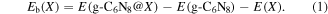
Here, Eb(X) is the binding energy of the dopant. E(g-C6N8@X), E(g-C6N8) and E(X) are the total energies of g-C6N8 doped with TMs, pristine g-C6N8 and isolated dopant, respectively.
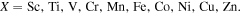
The following relation can calculate binding energies of CO2 molecules on g-C6N8@TMs
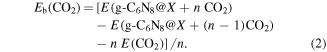
In equation (2), the first and second terms represent the total energies of TM functionalized g-C6N8 loaded with n and (n − 1) CO2 molecules respectively, where n is the number and E (CO2) is the total energy of CO2 molecule.
Results and discussion
Structural properties of g-C6N8 and g-C6N8@TMs
Before studying the CO2 storage properties of g-C6N8@TMs, we will briefly describe the structural properties of the monolayers in their pristine and functionalized form. The optimized C–N bond lengths of the atoms that constitute the six member rings and the atoms that joined the rings are found to be 1.476 Å and 1.33 Å, respectively. The lattice parameter after relaxation is 7.132 Å. These values agree well with the literature [30]. The lowest energy configuration of a 2 × 2 supercell of g-C6N8 is given in figure 1.

Figure 1. Optimized structures of g-C6N8. Gray and blue balls represent C and N atoms respectively.
Download figure:
Standard image High-resolution imageIn its pure form, g-C6N8 is inert towards CO2, thus to enhance g-C6N8–CO2 interaction to an acceptable value for ambient condition CO2 storage, we introduced selected TMs on different binding cites available in g-C6N8. A set of selected TMs from Sc–Zn has been considered for the doping process. While considering the suitability of the doping mechanism of foreign adatoms on 2D g-C6N8 monolayers, the most important parameters are stability and reversibility of the doped systems. Both of these parameters could be established through the determination of the Eb/Ec ratio [38–42]. For Eb/Ec > 1, a uniform scattering of dopants over the g-C6N8 is preferred that ensures stability and reversibility, whereas Eb/Ec < 1 would lead towards cluster formation instead of binding. Considering this fact, we have carefully calculated the Eb/Ec ratio for the selected TMs over g-C6N8.
For convenience we will explain here the case of Sc on g-C6N8. In order to find the most stable binding site, we introduced Sc on all the possible positions, which are C/N top, bridge and hollow sites formed by a small ring, a bridge of atoms linking the small rings and a big pore. The Sc dopant, similar to all the other considered TMs dopants, prefers to sit in plane with g-C6N8 in the big pore with significantly high binding energies (Eb) as shown in figure 2.

Figure 2. Optimized structures of selected TM functionalized g-C6N8. Gray and blue balls represent C and N atoms, respectively, where other TMs dopants are represented by the colors as mentioned. All the TMs prefer to sit in the big pore of g-C6N8.
Download figure:
Standard image High-resolution imageIn the case of Sc, the calculated Eb value of −12.21 eV is much higher than its experimental Ec and Sc sits exactly in the center of the big pore. An average distance of Sc with the six neighboring N atoms is 2.39 Å. After introducing Sc, the g-C6N8 monolayer does not remain exactly planar but crumbled a little. However this slight crumbling is reversible, not permanent, and the monolayer comes back to its planar form once Sc dopant is removed and the systems is optimized again. With Eb/Ec ∼ 3.13, Sc would stay on g-C6N8 comfortably even at significantly high temperature and nullifies the likelihood of cluster formation. Other TM dopants also ensured Eb/Ec > 1, for example Ti ∼ 2.6, V ∼ 2.6, Cr ∼ 3.9 and Mn ∼ 5.3, thus guaranteeing the stability and reversibility of the functionalized systems. It is worth mentioning here that the binding mechanism and Eb values of the considered TMs on g-C6N8 follow a similar trend to that of g-CN [43]. A comparative analysis of Eb to Ec of the studied TMs is given in figure 3.
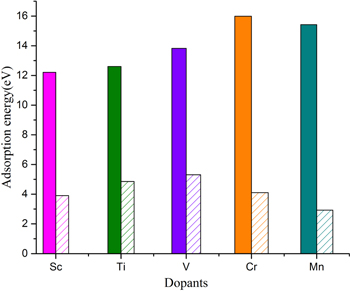
Figure 3. van der Waals corrected Eb (solid colored bars) versus experimental Ec of selected TMs doped with g-C6N8. For convenience in plotting, we have considered Eb and Ec values as positive.
Download figure:
Standard image High-resolution imageA strong Eb is evidence of the fact that a significant amount of charge is being transferred from the TMs (with smaller electronegativities) to the N atoms (with higher electronegativity) of g-C6N8, with which dopants are bonded. Let us consider the case of Sc–g-C6N8; Sc loses an electronic charge of around 1.701 e to its closest N atoms of g-C6N8. This depletion and accumulation of charges can be seen in the isosurface charge density plots in figure 4. Other TM dopants also acquire partial positive state upon losing a substantial amount of their electronic charges to their nearby N atoms of g-C6N8. This transfer of charge would also result into a strong hybridization between the TMs and g-C6N8, which is discussed in the electronic properties section.
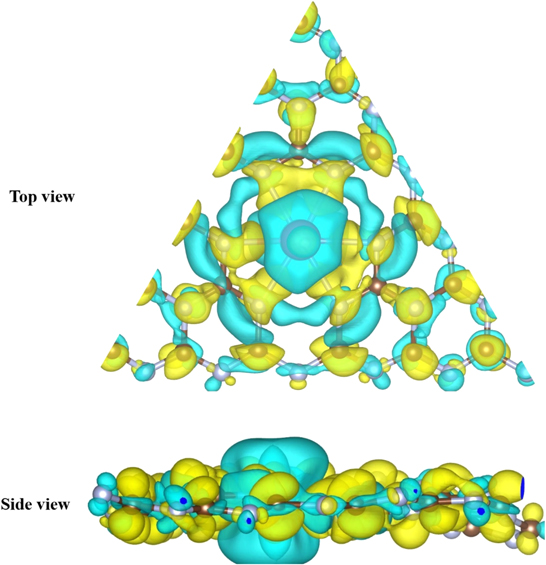
Figure 4. Isosurface charge density of g-C6N8–Sc. Here, yellow and cyan colors indicate the accumulation and depletion of charges, respectively. We have used isovalue = 0.005 e Å−3.
Download figure:
Standard image High-resolution imageAdsorption mechanism of CO2 on g-C6N8@TMs
After establishing a strong TMs-g-C6N8 binding, we introduced multiple CO2 molecules on these functionalized systems one by one. It is worth mentioning here that after the Tawfik et al study of CO2 storage by defected graphene doped TMs [16], the present study is the only attempt to report the storage of several CO2 on TM functionalized g-C6N8. All the doped systems anchor CO2 under a similar mechanism; however, as a representative system, we will explain the case of Sc@g-C6N8. For CO2 adsorption, we have tried several adsorption configurations of CO2 on Sc@g-C6N8 by considering different binding distances of CO2 from cationic Sc+ as well as by changing the orientations of the incident CO2 molecules like C-directed, O-directed and tilted molecule. Energetics analysis all the CO2-Sc@g-C6N8 configurations reveal the most stable structure, as shown in figure 5.
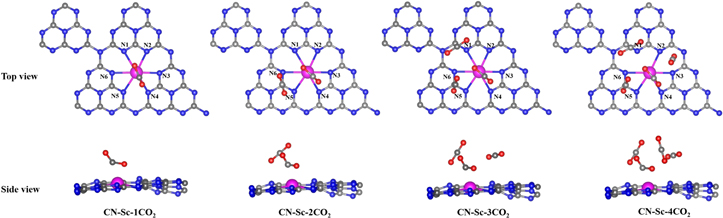
Figure 5. Top and side views of the optimized structures of selected multiple CO2 molecules on Sc-g-C6N8. Gray, blue, magenta and red balls represent C, N, Sc and O atoms, respectively.
Download figure:
Standard image High-resolution imageThe lowest energy configuration of single CO2 on Sc-g-C6N8 yields an Eb value of −0.77 eV at a binding distance of 2.07 Å, which agrees very well with the single CO2 adsorption on Ca and selected TM doped carbon-based nanostructures [16, 18, 44]. This reasonably high Eb and appropriate binding distance suggest that Sc-g-C6N8 is a good option for reversible CO2 capture. Upon CO2 adsorption, Sc-g-C6N8 crumbles a little further and a CO2 molecule also rearranges its structure with the elongation of C–O bond lengths to 1.21 Å and 1.29 Å, from an equilibrium value of 1.16 Å. The molecule also loses its planar geometry with the shrinking of the O–C–O angle to 136.4°. Now we study the coverage effect by introducing more CO2 to Sc-g-C6N8, which is pre-adsorbed with single CO2. While studying the multiple CO2 storage mechanism, we adopted the similar principle regarding binding energies and binding distances as that of [16]. We kept on increasing the number of CO2 on Sc-g-C6N8 as long as the resulting Eb values exceeded a threshold of −0.25 eV/CO2 and the binding distance remained lower than 4.0 Å, which are the usual physisorption parameters [45]. The second CO2 molecule was introduced by considering several configurations and maintaining a suitable distance not only with Sc-g-C6N8 but also with the pre-adsorbed CO2, to avoid any possible repulsion between CO2 molecules. A resultant ground state configuration yields average Eb and binding distance of −0.50 eV/CO2 and 3.0 Å respectively. Upon structural relaxation, there is hardly any change in the geometry of the second CO2, which has C–O bond lengths of 1.18 Å each. Now for the adsorption of the third CO2, we considered the ground state configuration of Sc-g-C6N8 with 2CO2 already adsorbed. Most stable configurations yield Eb values of −0.42 eV and −0.37 eV per CO2 in the case of 3CO2 and 4CO2 on Sc-g-C6N8, respectively. Adsorption distances in all the cases up to 4CO2 fall within the prescribed criteria mentioned above. However the adsorption of the fifth CO2 on Sc-g-C6N8 pre-adsorbed with 4CO2 would not only result into an Eb that is smaller than −0.25 eV/CO2 but also at a larger distance, falling short to the laid down criteria. It is therefore concluded that Sc-g-C6N8 could take 4CO2 with reasonable Eb and binding distances. Optimized structures of Sc-g-C6N8 with multiple CO2 are shown in figure 5. Similar to Sc-g-C6N8, we have studied the mechanism of multiple CO2 storage on g-C6N8 functionalized with other TMs and results of the respective Eb are given in figure 6. It is clear from figure 6 that with the increasing number of CO2, the Eb decreases due the increase of distances from CO2 and Sc-g-C6N8 and possible repulsion among the partially polarized pre-adsorbed CO2.

Figure 6. van der Waals corrected Eb values of selected TM doped Sc-g-C6N8.
Download figure:
Standard image High-resolution imageElectronic properties
The analysis of DOS, shown in figure 7(a), indicates that g-C6N8 is semiconducting where its valence band and conduction band are predominantly formed by N-2p and the overlap of N-2p and C-2p, respectively. In particular, the remarkable overlap of N-2p and C-2p accounts for a sigma C–N bond (i.e., 0.5–2.5 eV as anti-bonding and −6.0 to −4.0 eV as bonding). This resembles the character of photocatalytic material g-C3N4. The integration of a Sc atom into the hole of g-C6N8 results in the significant shift in the Fermi level to the conduction band, manifesting the n-type impurity that originates from the donation of two outermost 4 s electrons of Sc to the π* state of the sheet [43]. As a consequence, the donating atom has become a positively charged Sc2+ ion of which there is a localized electron in the 3d orbital (d1) as evidenced by the major spin-up close to the Fermi level (figure 7(b)). According to the optimized structure of Sc-doped C6N8, six N atoms surround the Sc dopant. (i.e., the hexagonal crystal field) This field leads to splitting of the d orbital where the d1 electron is filled in the lowest degenerate states either dxz or dyz [43]. Therefore, it is prone to interact with adsorbates (here CO2 molecules) via orbital overlapping.

Figure 7. Density of states (DOS) of (a) g-C6N8 and (b) g-C6N8 adsorbed by a Sc atom. The Fermi energy is shifted to zero for the sake of convenient visualization and the arrow denotes spin-up and spin-down.
Download figure:
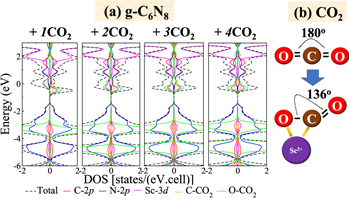
Standard image High-resolution imageFigure 8(a) shows the DOS Sc-doped C6N8 adsorbed with various numbers of CO2 molecules. There is the apparent overlap of Sc 3d with the LUMO (mainly derived from C and O 2p orbitals at around −0.5 to 0.5 eV) of the first CO2 molecule. This overlap is responsible for the formation of strong covalent Sc–C and Sc–O bonds. As a result, they notably induce two major modifications (1) the elongation of two C–O double bonds in CO2 from the initial value of 1.16 Å to 1.29 Å and 1.21 Å and (2) the bending of C–O–C from 180.0° to 136.4°, as schematically depicted in figure 8(b). The subsequent adsorption of the second CO2 leads to the gradual disappearance of magnetic moment due to delocalization. Interestingly, further inclusion of the third and fourth CO2 molecules yield negligible changes in the DOS, indicating that they are weakly bound to the functionalized sheet by van der Waals forces with the probable adsorption energy as described previously. The presence of the van der Waals interaction of the second to fourth molecules is also supported by the slight changes in their C–O bond length and C–O–C bond angle. Therefore, it is conclusive that a Sc-doped C6N8 sheet is a suitable material for CO2 storage with exceptional storing capacity.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. DOS of (a) Sc-doped C6N8 adsorbed by CO2 molecules and (b) the schematic illustration of bond bending of the first adsorbed CO2 molecule.
Download figure:
Standard image High-resolution image{kind=link}
Conclusion
In conclusion, we have performed rigorous ab initio calculations based on DFT, to design and investigate metallized g-C6N8 monolayers as potential CO2 adsorbents for environment protection. A set of selected TMs that includes Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu and Zn have been functionalized on a supercell of g-C6N8 at a doping concentration of 1.79%. Our van der Waals induced calculations have revealed that each dopant sticks to a g-C6N8 nano sheet without forming metal clusters, which is extremely important for the reversibility of the doped system. The doping mechanism involves a transfer of electronic charge from TMs to the g-C6N8, altering the electronic properties of the latter without compromising its stability. Each TM's dopant in cationic form is capable of anchoring a maximum of four CO2 molecules with binding energies, which are much higher than that of pristine g-C6N8. We strongly believe that these findings will provide the basis for the synthesis of a promising class of functional nanostructures with application in feasible CO2 storage.
Acknowledgments
T H is indebted to the resources at NCI National Facility systems at the Australian National University through National Computational Merit Allocation Scheme supported by the Australian Government and the University of Queensland Research Computing Centre. R A acknowledges the Swedish Research Council (VR), Carl Tryggers Stiftelse för Vetenskaplig Forskning and StandUp for financial support. The SNIC and UPPMAX are also acknowledged for provided computing time. T K would like to acknowledge the Development and Promotion of Science and Technology Talent Project (DPST) for the financial support of this project. The Nanotechnology Centre (NANOTEC), NSTDA Ministry of Science and Technology (Thailand) also supports T K through its program of Centre of Excellence Network, Integrated Nanotechnology Research Centre Khon Kaen University (Thailand).