
Abstract
The sensitive nature of molecular hydrogen (H2) interaction with the surfaces of pristine and functionalized nanostructures, especially two-dimensional materials, has been a subject of debate for a while now. An accurate approximation of the H2 adsorption mechanism has vital significance for fields such as H2 storage applications. Owing to the importance of this issue, we have performed a comprehensive density functional theory (DFT) study by means of several different approximations to investigate the structural, electronic, charge transfer and energy storage properties of pristine and functionalized graphdiyne (GDY) nanosheets. The dopants considered here include the light metals Li, Na, K, Ca, Sc and Ti, which have a uniform distribution over GDY even at high doping concentration due to their strong binding and charge transfer mechanism. Upon 11% of metal functionalization, GDY changes into a metallic state from being a small band-gap semiconductor. Such situations turn the dopants to a partial positive state, which is favorable for adsorption of H2 molecules. The adsorption mechanism of H2 on GDY has been studied and compared by different methods like generalized gradient approximation, van der Waals density functional and DFT-D3 functionals. It has been established that each functionalized system anchors multiple H2 molecules with adsorption energies that fall into a suitable range regardless of the functional used for approximations. A significantly high H2 storage capacity would guarantee that light metal-doped GDY nanosheets could serve as efficient and reversible H2 storage materials.
Export citation and abstract BibTeX RIS
Introduction
With the rapid exhaustion of fossil fuel and an ever-growing energy demand, it is now impractical with our limited fossil fuel reserve to meet the expectations of modern society. The prospect of hydrogen-based fuel-cell technologies as an alternative, renewable and sustainable source of clean energy has been pursued for a long time. Although, as an energy carrier, hydrogen (H2) is efficient, abundant and environmentally benign, storing H2 is still a vital challenge. A lot of research efforts have been devoted towards developing suitable H2 storage materials with high gravimetric and volumetric densities, and at the same time ensuring energy-efficient H2 release at the time of utilization [1, 2].
In this context, carbon-based two-dimensional (2D) nanostructured materials have attracted a great deal of attention as high-capacity H2 storage materials capable of meeting the US Department of Energy (DOE) target of 9 wt % [3–6]. Graphene, a pioneer of 2D materials, which has a large surface area coupled with the existence of Dirac cones, has huge potential for applications as an efficient H2 storage material [7, 8]. The binding of H2 on pristine graphene is a weak van der Waals-type interaction, which is not suitable for ambient condition applications. This weak binding could further be enhanced by means of defect formation, foreign atom doping or using other graphene-like 2D materials. Recently, several attempts were made to increase the binding of H2 by using metal-doped hydrogenated and fluorinated graphene sheets called graphane and flouro-graphene, respectively [9, 10]. There have been further efforts to decorate various 2D materials including graphene with alkali metals (AMs), alkaline earth metals (AEMs) and transition metals (TMs). Each dopant (AM, AEM, TM) on 2D materials can bind several H2 molecules resulting in sufficient H2 storage capacity. Compared to AMs or AEMs, in the case of TM doping, the enhanced binding of H2 is due to the Kubas interaction, which results from the hybridization of metal d states and the bonding/anti-bonding orbitals (σ and σ*) of H2 [11–22].
For reversible H2 storage, the current focus is to look for a carbon-based material with larger pore size, not only for H2 but also for the dopants used for functionalization. In this regard, graphdiyne (GDY), a new carbon allotrope, has been successfully synthesized and predicted as the most stable diacetylenic carbon allotrope [23]. GDY, composed of a lattice of benzene rings with sp and sp2 hybridized carbon atoms, has much larger pores than the densely packed honeycomb lattice structures of graphite and graphene. The successful fabrications of GDY monolayer report a natural band-gap opening of nearly 0.46 eV, unlike zero band-gap graphene, due to the asymmetric π bindings and with high electron mobility of 105 cm2 V−1 s−1 at room temperature similar to that of graphene at room temperature conditions [24]. Therefore, GDY sheets with larger nanopores could be an attractive candidate for various applications including H2 storage. Zhang et al synthesized GDY by cross-coupling reaction and further studied its magnetic properties, concluding that GDY is paramagnetic [25]. By means of density functional theory (DFT) calculations, Chen et al studied the catalytic ability of GDY supported by a Ag38 cluster for CO oxidation and found it very promising [26]. In a recent experimental study describing the application of GDY, Lv et al conducted a reaction of a metal-free catalyst in oxygen reduction [27]. Hongyu et al used DFT to study the intercalation and diffusion of H2 in single-layer and bulk GDY, which is the guiding point for understanding the H2 storage capacity of GDY [28].
In addition to the few reports mentioned above, GDY is promising in various technological applications and has been a subject of many studies, however its use as H2 storage medium is less explored. Bridging this gap, we have performed a comprehensive DFT study to investigate the potential of light metal functionalized GDY in H2 storage applications. In this regard, the interaction of H2 molecules with those of GDY or dopants is very crucial and should be treated very carefully. Usually, H2 binds with pristine surfaces under weak van der Waals (vdW)-type forces, which could be taken care of by adding a correction term to frequently used functionals like generalized gradient approximation (GGA) with Perdew, Burke and Ernzerhof (PBE). However, the contributions of non-local long-ranged vdW forces has never been considered in case of H2 storage materials. Though for largely homogeneous system, and for inhomogeneous systems, the GGA works well within DFT, for sparse systems including gas molecules, biomolecules, having inter-particle separations, the non-local, long-ranged vdW forces are influential. The non-local vdW density functional (vdW-DF) by Dion et al [29] is a very promising scheme for the efficient treatment of weakly bonded systems. The rigorous vdW-DF method can dramatically improved the electronic structure of dispersion, which often occurs in conjunction with hydrogen bonds. Thus, the present study is the first attempt to consider the contributions of long-ranged vdW forces in addition to commonly used DFT-D3.
Here we have studied the H2 storage property of a GDY sheet decorated with different light metals such as Li, Na, K, Ca, Sc and Ti using a first-principles DFT calculation with DFT-D3 [30] correction and with non-local vdW-DF [31]. We find all the dopants bind strongly the to GDY sheet with Sc and Ti binding more strongly as compared to Li, Na, K and Ca. Each functionalized system has high affinity towards H2 molecules with an average binding energy per H2 molecule falling within a desirable range for particle applications. The unique feature of this study is the comparison of H2 adsorption energies calculated by GGA, vdW-DF and DFT-D3.
Method and computational details
For this work, spin-polarized DFT simulations were performed by using the Vienna ab initio simulation package (VASP) [32, 33]. The GGA-PBE [34] was used for exchange–correlation energy within the projector-augmented wave (PAW) method [35]. It is well known that the normal DFT fails to describe interactions at long range, such as vdW interactions within semi-LDA or with gradient-corrected GGA approximation. To include vdW interactions, we have included the very robust DFT-D3 approach by Grimme [36], which has been the most widely used approach to the dispersion problems. We have also implemented non-local correlation vdW-DF in our calculations and compared our results.
Here, on one side, the effect of vdW interactions is included explicitly by using the empirical correction scheme of Grimme (DFT +3). DFT-D3 is a vigorous method, which has successfully been applied to address intermolecular dispersion correction in different cases like the interaction of various gas molecules with 2D graphene-like sheets. This approach is less empirical and gives better accuracy for lighter molecules and a strongly improved description for heavier systems.
For DFT-D3, total energy is calculated as,
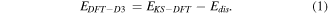
Here, Eks−DFT is the self-consistent Kohn–Sham energy obtained from the DFT calculation and Edis is the dispersion correction. However, the most serious drawback with the DFT-D3 approach is that the correction is not dependent on the electronic structure.
On the other side, in vdW-DF the non-local correlation, the exchange–correlation energy is calculated as described below
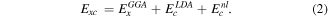
Here,  is the exchange energy calculated with the PBE functional.
is the exchange energy calculated with the PBE functional.  is the local correlation energy within the LDA approximation.
is the local correlation energy within the LDA approximation.  is the non-local correlation energy.
is the non-local correlation energy.
Our model GDY system contains 18 C atoms. In the super cell geometry, to avoid the spurious interactions between neighboring periodic structures, a vacuum of 20 Å was taken along the non-periodic z direction (perpendicular to the plane of GDY). For the ground-state optimization of the GDY as well as metal decorated GDYs, SCFs were carried out until the Hellmann−Feynman force acting on each atom was less than 0.001 eV Å−1. The energy convergence criterion was chosen to be 10−7 eV per atom and plane-wave basis set cut-off was chosen to be 500 eV. The Brillouin zone integration was performed by the Monkhorst−Pack scheme [37] with a 3 × 3 × 1 k-point sampling for the structure optimization and a denser sampling of 5 × 5 × 1 for energy optimization. Spin-polarization is included throughout the calculations.
The binding energy Eb of metal decorated GDY sheets (GDY-M) is calculated as,
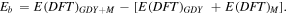
Here, E(DFT)GDY+M is the KS-DFT energy of the metal decorated GDY sheet. E(DFT)GDY and E(DFT)M are the KS-DFT energy of bare GDY sheet and isolated metal atoms (M = Ti, Sc, Li, Na, K, Ca), respectively.
Results and discussion
First, we will discuss the structural properties of pristine and metal functionalized GDY. As mentioned above, GDY possesses large periodic pores uniformly distributed and connected with hexagonal rings as shown in the optimized stricture shown in figure 1.

Figure 1. Top (a) and side (b) view of 18 carbon (brown) atoms GDY sheet. The dotted line (magenta) marks the primitive of GDY with a small pore (S) and a large pore (B).
Download figure:
Standard image High-resolution imageThe optimized C–C bond length of the C atoms that constitutes small and large pores is found to be 1.43 and 1.39 Å, respectively.
Now we discuss the doping mechanism of GDY with various light metal dopants. Several doping cites available in GDY have been considered to find the lowest energy configurations. We have studied different configurations for functionalizing the GDY sheet with the M atoms. First, we have calculated the adsorption energy of a single M atom on available symmetry site of one side of GDY sheet. From the five possible adsorption sites, large pore top (B), small pore top (S), carbon top on small pore (C1), carbon top on large pore (C2) and bridge top (B1), the large pore B site is found to be most suitable for the metal decoration. Our DFT-D3 calculated binding energies of Li, Na, K, Ca, Sc and Ti has been found to be −4.31 eV, −3.45 eV, −4.30 eV, −3.06 eV, −5.19 eV and −3.73 eV, respectively. These high bindings encourage us to enhance the doping concentration of light metal dopants on GDY. We took the optimized geometries of single dopant GDY systems as an initial structure and tried all possibilities for the second metal atom to be accommodated. Our rigorous computations revealed that a large pore is the most stable site for all the metal dopants considered here. The optimized structures of metal (M) decorated with 2Li, 2Na, 2K, 2Ca, 2Sc and 2Ti on GDY sheets are depicted in figures 2(a)–(f). The average binding energies per dopant in case of two metal atoms on GDY has been found as −2.28 eV, −2.00 eV, −2.39 eV, −3.23 eV, −4.93 eV and −5.23 eV for Li, Na, K, Ca, Sc and Ti, respectively. The minimum metal–C of GDY distances is C–Li (2.21 Å), C–Na (2.81 Å), C–K (2.96 Å), C–Ca (1.41 Å), (a), C–Sc (2.16 Å) and C–Ti (2.09 Å). The optimized distance between Ti–Ti, Sc–Sc, Li–Li, Na–Na, K–K and Ca–Ca are calculated to be 4.88, 4.75, 4.76, 5.46, 5.48 and 3.78 Å, respectively. The distances between the functionalized atoms coupled with strong binding energies are large enough to avoid metal–metal clustering on the GDY sheet.

Figure 2. Optimized structure of metal decorated GDY sheets functionalized with (a) Ti (cyan blue), (b) Sc (purple), (c) Li (green), (d) Na (yellow), (e) K (violet) and (f) Ca (blue).
Download figure:
Standard image High-resolution imageElectronic structure and charge analysis
The varying binding energies between the studied metal atoms on the GDY sheet is well explored by studying the electronic density of state (DOS) of the functionalized system along with the charge transfer mechanism. The spin-polarized DOS plot of GDY in figure 3(a) depicts the semiconducting behavior of the sheet with a band-gap of 0.2 eV. From the spin-polarized partial density of state (PDOS) plots, it is evident that in case of Sc functionalized GDY (Sc-GDY), there is a strong hybridization between Sc(3d) and Sc(4s) with C(p) of CNT states near the Fermi level, as shown in figure 3(b). Whereas, for Ti-functionalized GDY (Ti-GDY), the strong hybridization between Ti(4d) and Ti(4s) with that of C(p) states is evident near the Fermi level, as shown in figure 3(c). Whereas, in the case of Li-functionalized GDY (Li-GDY), K- and Na-functionalized GDY (K-GDY and Na-GDY), Li(2s), shows small hybridization, in the case of Na(3s), K(4s), the hybridization between the C(p) states of CNT are very weak as evident near the Fermi energy in figures 3(d)–(f).

Figure 3. Spin-polarized density of states of a pristine GDY sheet (a). Spin-polarized partial density of states of (b) Sc-, (c) Ti-, (d) Li-, (e) Na- and (f) K-functionalized GDY, respectively.
Download figure:
Standard image High-resolution imageTo verify the charge transfer mechanism, we have performed a Bader-charge analysis. From the Bader analysis, we found a redistribution of charge among the C atoms in the GDY structure. The C atoms in the small benzene S-ring in figure 1 have a smaller charge density (2.5 e) compared to the C atoms associated with the larger pore (6.2 e) of GDY (B-ring in figure 1). With Sc doping, both of the Sc atoms lose a significant amount of charge of 2.04e and become cationic. The charge from Sc gets accumulated on the C atoms of the S-ring and on the C atoms laying in close proximity of the Sc atoms in the B-ring. In the case of Ti-doped GDY, each Ti atom loses a charge of 1.96 e whereas, the C atoms have an increased charge density. Whereas, in the Li-GDY, K-GDY and Na-GDY sheets, Li, K and Na, although weakly bound, lose almost 90% of their charge (0.9e) and become cationic. The C-GDYs, near the functionalized metal atoms mostly accumulate that charge. The depletion and accumulation of electronic charges in the case of Ti-, Li- and Ca-doped GDY can be seen in figure 4.
Hydrogenation
In general, the adsorption of H2 on the surfaces of nanostructures, especially pristine and functionalized 2D materials, is very sensitive and mostly the interaction is of weak vdW type. Thus, to calculate the H2 adsorption energy, we have carefully chosen the vdW functional prescribed in different formats in our DFT calculation and compared our results. In one method, we have explicitly included the empirical correction scheme of Grimme (DFT + D3) as described in equation (1) and, in the second method, we have implemented the vdW-DF non-local correlation where the exchange–correlation energy is calculated as described in equation (2). The H2 adsorption energies of both DFT-D3 and vdW-DF have been further compared with GGA functionals, which does not consider any vdW corrections.
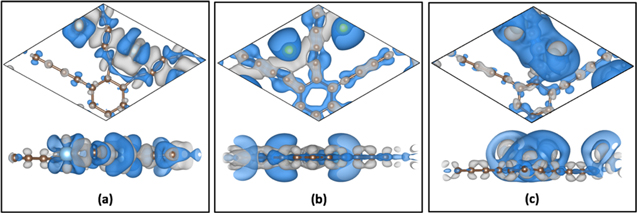
Figure 4. Isosurface charge density of (a) Ti-, (b) Li- and (c) Ca-functionalized GDY nanosheets. Gray and blue regions depict accumulation and depletion of charges.
Download figure:
Standard image High-resolution imageAfter having the optimized structures of the functionalized GDY sheets with 11% light metals coverage, each of the 2Sc-GDY, 2Ti-GDY, 2Li-GDY, 2Na-GDY, 2K-GDY and 2Ca-GDY sheets were exposed to H2 molecules. The H2 molecules were introduced at different possible configurations near the functionalized M atoms. At each configuration, the hydrogenated M-GDY sheets (M-GDY-nH2) were optimized to obtain the ground-state configuration and the energies were calculated as,
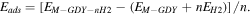
Here, Eads is the H2 adsorption energy of the hydrogenated metal functionalized GDY sheets. EM−GDY−nH2 is the total energy of hydrogenated metal functionalized GDY sheets (M = Sc, Ti, Li, Na, K). EM−GDY and EH2 are the total energies of metal functionalized GDY sheets and H2 molecule, respectively. Here, n is the number of H2 molecules.
Following optimization of the M-GDY sheet with four H2 molecules (each M atom exposed to two H2 molecules), the number of H2 molecules around each M atom was gradually increased to the maximum adsorption limit, which is four H2 molecules per dopant. The top and side views of the lowest energy configuration of hydrogenated systems with 4H2, 6H2 and 8H2 molecules are shown in figures 5(a)–(c), respectively. The adsorption energy per H2 molecule calculated by GGA, vdW-DF and DFT-D3 on M-GDY sheets is shown in figures 6(a)–(c), respectively.

Download figure:
Standard image High-resolution image
Download figure:
Standard image High-resolution image
Figure 5. (a) Optimized structures of (a) GDY-2Ti, (b) GDY-2Sc, (c) GDY-2Li, (d) GDY-2K, (e) GDY-2Ca and (f) GDY-2Na saturated with 4H2 each. (b) Optimized structures of (a) GDY-2Ti, (b) GDY-2Sc, (c) GDY-2Li, (d) GDY-2Na, (e) GDY-2K and (f) GDY-2Ca saturated with 6H2 each. (c) Optimized structures of (a) GDY-2Ti, (b) GDY-2Sc, (c) GDY-2Li, (d) GDY-2Na, (e) GDY-2K and (f) GDY-2Ca saturated with 6H2 each.
Download figure:
Standard image High-resolution image
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. Average adsorption energies per H2 for 4H2, 6H2 and 8H2 on metal functionalized GDY calculated by (a) GGA, (b) vdW-DF and (c) DFT-D3.
Download figure:
Standard image High-resolution image{kind=link}
Under maximum H2 coverage, which is 8H2 molecules on M-functionalized GDY systems, the average adsorption energies per H2 in the cases of GGA, vdW-DF and DFT-D3 have been found to be within the range of −0.10 to −0.197 eV, −0.194 to −0.173 eV and −0.345 eV, respectively. From a comparative analysis of the H2 adsorption energies calculated by different functionals considered in this study we conclude a trend GGA > vdW-DF > DFT-D3, which is consistent with previous studies [38]. Regardless of the functional used in calculations, all of these H2 adsorption energies fall within the desirable range for the application of an efficient H2 storage medium under practical conditions.
The H2 storage capacity (gravimetric density) H2(G) can be estimated as
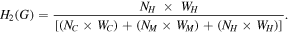
Here, N represents the number of each type of atoms in the M–GDY–H system (C, (M = Li, Na, K, Ca, Sc and Ti) and H) and W represents their corresponding molecular weight.
Using the above relation, reasonably high H2 storage capacities of 6.50, 5.80, 5.20, 5.13, 5.0, and 4.91 wt% have been achieved for Li, Na, K, Ca, Sc and Ti, respectively, with only 11% metal doping concentrations. Increasing the metal doping in GDY nanosheets, H2 storage could be further enhanced.
Conclusion
In this study we have taken up the issue of the debatable adsorption mechanism of H2 with pristine and functionalized 2D nanostructures though a specific example of GDY nanosheets. We have employed DFT calculations by means of different approximations namely GGA (without adding any corrections), vdW-DF (non-local correlation) and DFT-D3 (vdW corrections). In addition to investigating the structural and doping mechanism of GDY with light metals like Li, Na, K, Ca, Sc and Ti, their electronic, charge transfer and H2 storage characteristics have also been explored. It is found that each dopant binds strongly with GDY at a doping concentration of 11% and transforms the electronic properties of the latter from semiconductor to metallic. The metal–GDY bonding turns the dopants into cations, which is essential for the adsorption of H2 molecules. Each functionalized GDY system anchors eight H2 molecules with H2 adsorption energies within an acceptable range regardless of the functional used for the approximations. Two dopants per GDY yielded a high storage capacity of 6.50 wt%, which could further be enhanced through high metal doping. Our results revealed that light metal-doped GDY nanosheets are attractive H2 storage materials with a high H2 storage capacity and ideal adsorption energies.
Acknowledgments
We are indebted to the CENCON, Swedish Research Council (VR), Carl Tryggers Stiftelse för Vetenskaplig Forskning for financial support. The SNIC and UPPMAX are acknowledged for providing computing time. We are thankful to UQ for providing the financial support and the resources at the NCI National Facility systems at the Australian National University through the National Computational Merit Allocation Scheme supported by the Australian Government and the University of Queensland Research Computing Centre.