

Abstract
We present the 0 K structures and formation energies for vacancy clusters of up to four vacancies and migration barriers for a single vacancy at a semicoherent Kurdjumov–Sachs Cu–Nb interface using ab initio calculations. Two main results emerge from this study, first that the predicted vacancy structure is compact, differing notoriously with predictions based on available empirical potentials, and second that vacancy clusters containing up to four vacancies have a smaller formation energy than monovacancy in bulk. Additionally, the binding energies show that the vacancy clusters are energetically stable for clusters having up to four vacancies. Nudged elastic band calculations of migration barriers show that the migration of a vacancy from one misfit dislocation intersection to another is highly improbable due to the high barriers. These findings suggest that at nonzero temperatures the interface will be preloaded with vacancy clusters with a relatively large capture radius for interstitials in the interface plane, implying that the semicoherent Cu–Nb interface could be a highly effective sink for point defects that form due to irradiation.
Export citation and abstract BibTeX RIS
1. Introduction
The irradiation induced changes in properties of materials such as dimensions, ductility, hardness, corrosion resistance, etc are ultimately either directly caused or mediated by formation of point defects. By controlling the effective rate at which the point defects are produced, many of the undesirable effects such as swelling or embrittlement could be prevented or minimized. It has been shown that in materials with a special kind of nanostructure, namely with alternating immiscible fcc-bcc metallic layers the interfacial planes, can act as highly efficient sinks for point defects and be morphologically stable under irradiation [1].
Cu–Nb nanoscale metallic multilayer composites (NMMCs) have been thoroughly studied both experimentally [2–4] and theoretically [5, 6]. For example, a 20 nm thick void-denuded zone parallel to the interfacial plane has been observed in experiments, attributed to the emission of interstitials from the interface and subsequent capture and annihilation of vacancies [7]. Atomistic simulations based on density-functional theory (DFT), molecular dynamics (MD) and several flavors of Monte Carlo (MC) allow for atomic scale resolution investigations of structure, point defect creation, migration and clustering, usually providing more information than accessible via current experimental methods. Given proper models it would be possible to predict and optimize the properties of materials for specific applications.
Despite DFT-based calculations having become standard in computational materials science, only a few studies using this method have been performed for semicoherent metal–metal interfaces [8–10]. These confirmed some of the earlier research done using empirical interatomic potentials (EIP), but also uncovered some deficiencies. For example, lower than bulk formation energies for point defects at misfit dislocation intersections (MDIs) were predicted both by DFT and EIP [6], however the vacancy structure, equilibrium concentration and migration barriers differ. Although, this does not automatically invalidate conclusions drawn from EIP-based calculations, it still suggests the need for more DFT calculations in order to both validate or invalidate EIP predictions and also provide further insight about the underlying mechanisms as well as additional input into fitting better interatomic potentials.
In this paper we study the energetics and geometry of vacancy clusters at the Cu–Nb semicoherent interface using DFT and compare it to results obtained using EIPs. We also look at the migration of a single vacancy at the interface using nudged elastic band (NEB) calculations.
2. Method of calculation
A Cu–Nb structure consisting of 216 Cu and 120 Nb atoms containing an interface is used in the calculations. The Cu and Nb slabs are joined in Kurdjumov–Sachs (KS) orientation, where the {111} Cu and {110} Nb surfaces are joined in a way that the Cu crystal  direction is parallel to
direction is parallel to  Nb. The size of the system in the directions parallel to the interface is chosen to contain exactly one unit of the periodic 2D misfit dislocation network. With periodic boundary conditions in the plane of the interface, the sample is the smallest that still represents an infinite interface. The Cu and Nb slabs are terminated in the perpendicular direction with a vacuum layer that is 16.5 Å thick. Further details on the initial interface geometry and the justifications on using it as well as additional calculation details can be found from our previous work [8].
Nb. The size of the system in the directions parallel to the interface is chosen to contain exactly one unit of the periodic 2D misfit dislocation network. With periodic boundary conditions in the plane of the interface, the sample is the smallest that still represents an infinite interface. The Cu and Nb slabs are terminated in the perpendicular direction with a vacuum layer that is 16.5 Å thick. Further details on the initial interface geometry and the justifications on using it as well as additional calculation details can be found from our previous work [8].
The DFT calculations were made with plane–wave pseudopotential code Vienna Ab initio Simulation Package (VASP) [11–14] using a GGA-PBE exchange-correlation functional [15] and pseudopotentials [16, 17] provided by the package. Vacancy clusters consisting of up to four vacancies were relaxed with a fixed simulation box size. A climbing image method was employed in the NEB calculations of migration barriers [18] and the migration paths investigated were the two paths from one MDI to another along two Cu  directions. The shorter path will be referred to as 1NN and the longer as 3NN. Figure 3(a) presents the pristine interface with the MDI at its center. In the NEB calculations the Cu and Nb slabs were not allowed to move with respect to each other.
directions. The shorter path will be referred to as 1NN and the longer as 3NN. Figure 3(a) presents the pristine interface with the MDI at its center. In the NEB calculations the Cu and Nb slabs were not allowed to move with respect to each other.
The MD simulations used for comparison were performed with two embedded atom type (EAM) [19] interatomic potentials using LAMMPS software package [20]. First, using a potential created by Demkowicz et al (EAM1) [21] that was fitted to reproduce the substitutional defect energy in Cu and Nb in their equilibrium phases and, additionally, fitted the bulk modulus and lattice constant of a hypothetical B2 structure. Second, using a potential created by Zhang et al (EAM2) [22] that was fitted to reproduce the enthalpy of mixing of random Cu–Nb alloys over the whole composition range.
The formation energies were calculated using equation (1), where E(N) is the total energy of the structure with N vacancies, E0 is the total energy of the pristine Cu–Nb interface and E0Cu is the cohesive energy of Cu in the FCC bulk.

It is possible to estimate the number concentration of clusters in the dilute limit with equation 2, where Ef is the formation energy of the cluster, kB is the Boltzmann constant, and T the temperature. N is the number of vacancies forming a cluster and α is related to the geometry, which in our case is one for all clusters as there is only one geometrically equivalent configuration for each MDI.

The binding energy is defined by equation (3), where Ef(N) is the formation energy of a vacancy cluster consisting of N vacancies, and quantifies the excess energy of joining an isolated vacancy at an MDI with a cluster of  vacancies. Therefore a positive value corresponds to the tendency for the isolated vacancy to precipitate into the cluster, while a negative value corresponds to the tendency to remain isolated.
vacancies. Therefore a positive value corresponds to the tendency for the isolated vacancy to precipitate into the cluster, while a negative value corresponds to the tendency to remain isolated.
3. Results and discussion
The formation energy of a single vacancy at the MDI of a KS interface predicted by DFT is 0.37 eV as shown in table 1, which is considerably lower than the corresponding value in bulk Cu. This indicates that excess vacancies in the Cu layers will have a driving force towards the interface and become trapped there. Furthermore, the lower formation energy shows that assuming similar entropic contributions the equilibrium vacancy concentration at non-zero temperatures will always be higher at the interface than in the bulk. Moreover, it is energetically preferable to create up to four vacancies at an interface instead of one in the bulk as can be seen on figure 1. During an irradiation process this lower formation energy region would drive the vacancies created in the cascade into the interface where they could recombine with the interstitials that migrated there during the initial steps of the cascade or with interstitials migrating from the bulk of the material.

Figure 1. Formation energies of creating vacancy clusters at the semicoherent Cu–Nb interface in Cu layer as calculated with DFT and interatomic potentials. The horizontal dashed line denotes the vacancy formation energy in bulk Cu as predicted by DFT, other lines are guides to the eye.
Download figure:
Standard image High-resolution imageTable 1. Formation energies of 2D vacancy clusters at the semicoherent Cu–Nb interface in the Cu layer. The last column represents the monovacancy formation energy in bulk Cu as predicted by the methods used and is added for comparison only. All energies are reported in units of eV.
| 1 vac | 2 vac | 3 vac | 4 vac | Bulk | |
|---|---|---|---|---|---|
| DFT | 0.37 | 0.41 | 0.62 | 1.02 | 1.17 |
| EAM1 | 0.18 | −0.37 | −0.28 | 0.19 | 1.26 |
| EAM2 | 0.72 | 0.90 | 1.31 | 1.89 | 1.27 |
Compared to DFT results the formation energies obtained by using the EAM1 potential show a different behavior. The two and three-vacancy clusters have a negative formation energy meaning that at 0 K the interface would be preloaded with such small clusters. EAM2 shows similar qualitative behavior as DFT predicting lower-than-bulk, but non-negative formation energies, however quantitatively the energies are considerably higher. It is crucial to know the accurate formation energies in order to predict the equilibrium concentration of vacancies due to the exponential relation. We must emphasize that the DFT energies calculated in this work might not represent the true formation energies in the dilute limit due to several approximations, like small simulation cell size, as described in [8], however the comparison between DFT and EIPs was made using the same small cell. In addition, the formation energy of a Cu vacancy using EAM2 in a 52398 atom cell is 0.76 eV and the formation energy per vacancy for 21 vacancies (corresponding to all MDIs) in the same cell is 0.79 eV/vacancy showing that the size effect due to the small supercell is relatively small.
The structure of a single vacancy as predicted by DFT is localized at the MDI as seen on figure 3(b) and therefore the vacancy migration mechanism could involve hopping of nearby atoms into the vacancy as is the case in the bulk material. This was observed in the MD simulations of vacancy diffusion using the EAM2 potential, and is contrary to the prediction of EAM1.
Creating a divacancy at the semicoherent interface at the MDI site has a formation energy of 0.41 eV. This energy is much lower than for a monovacancy in the bulk. Furthermore, the binding energy of a divacancy is about 0.3 eV as shown on figure 2. This would suggest that finding divacancies at the interface is more probable than monovacancies and it shows that a monovacancy is able to absorb additional vacancy from the bulk of the material. The divacancy cluster has a slightly larger effective area at the interfacial plane than in the bulk (figure 3(c)). This could increase the capture radius for interstitials. In comparison, the binding energies obtained from both EAM1 and EAM2 are markedly larger compared to DFT, being around 0.5 and 0.7 eV respectively. This difference results in a larger driving force of forming divacancy clusters at the interface.

Figure 2. Binding energies of creating vacancy clusters at the semicoherent Cu–Nb interface as calculated with DFT and interatomic potentials. The lines are guides to the eye.
Download figure:
Standard image High-resolution image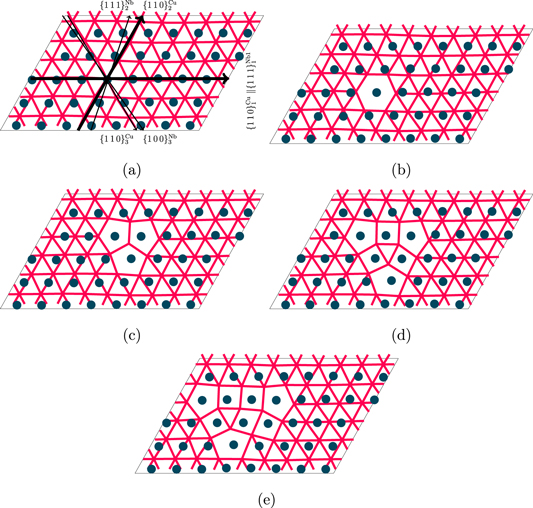
Figure 3. Relaxed structures of vacancy clusters at the Cu–Nb interface predicted by DFT calculations. Blue disks represent Nb atoms and the red lines show the bonds between Cu atoms (calculated with 3.3 Å cutoff), i.e. intersections of red lines show the locations of Cu atoms in the first layers next to the interface. (a) pristine interface, (b–e) interface with 1–4 vacancies.
Download figure:
Standard image High-resolution imageA similar reasoning as for the divacancy could be applied to the trivacancy as well since its binding energy is also positive. The area occupied by the defect cluster in the interfacial plane is larger (figure 3(d)) and therefore it is reasonable to assume that the capture radius for interstitials is also larger. Although the simulation box used is rather small for such a defect, the comparison simulations using the EAM2 potential with over 20000 atoms changed the formation energies by less than 0.05 eV. Therefore the results can be trusted qualitatively.
Finally, a tetravacancy cluster has a formation energy closer to the formation energy of a monovacancy in the bulk (table 1). The binding energy of forming a tetravacancy from a monovacancy and trivacancy is slightly negative, showing the limit of stable vacancy clusters that can form. In contrast to smaller clusters the geometry of the Cu layer at the interface gets more distorted and does not form a distinguishable structure (figure 3(e)).
The number concentration of vacancy clusters in MDIs at non-zero temperature can be estimated in the dilute limit from the acquired formation energies by using a simple exponential relation (equation (2)). At 600 K about 0.078% of MDIs are occupied with monovacancies, 0.071% with divacancies, and 0.002% with trivacancies. This result is many orders of magnitude larger than in the bulk Cu crystal per site. Therefore the concentration of vacancies in equilibrium is always higher than in bulk.
Since the growth of vacancy clusters at the interface is energetically favorable, it is also important to assess the probability of clustering from the kinetics perspective. In a previous DFT study [9] a single NEB calculation between two highest energy vacancy positions on the second shortest path between two MDIs was performed to assess the total effective migration barrier. In this study we calculated the full path including all local minima along the line as well as along the 1NN shortest path. The results are shown on figures 4 and 5. A distinctive feature of the vacancy migration behavior at these kind of interfaces appears where conventional migration barriers are modulated by the internal strain field of the interface. Whereas for the path between 1NN MDIs on figure 4 the barrier due to the strain field is higher, exceeding 1.0eV, the individual smaller barriers between two neighboring vacancy positions remain roughly the same throughout the whole path. This contrasts the 3NN MDI path where the barrier due to the strain field is lower, but the barriers between neighboring vacancy positions increase as the vacancy approaches the midpoint between the MDIs resulting in a similar overall effective migration barrier. In contrast, the NEB calculations done by Kolluri et al [23] using EAM1 potential show a markedly different behavior. It was found that the preferred migration takes place through a complex mechanism due to the delocalized monovacancy geometry. Above all, the calculated NEB landscape is complex and does not have the well structured oscillations as seen from our results. These results clearly indicate that vacancy structures and migration energies vary substantially according the the quality of the total energy model employed, which is the main result of this work.

Figure 4. The migration barriers for a vacancy moving along a straight path from an MDI towards the nearest MDI. The triangles correspond to the exact calculations, the solid line is a spline interpolation between the exact points and the dashed line is a spline interpolation between the local minima representing the increase in barriers due to the internal strain field. The energies are referenced to vacancy formation energy at an MDI.
Download figure:
Standard image High-resolution imageIt can be seen from figures 4 and 5 that hops to the nearest-neighbor sites around the MDI are highly probable at even low temperatures. This was seen in MD simulations with a large box using EAM2 at 500 K. Most of the hops occurred along the pathway between 3NN MDIs. On the contrary, the diffusion of a single vacancy into the next MDI has a low probability due to many hops needed along a pathway and due to the increasing barriers. Furthermore, it shows that breaking up of the vacancy clusters into individual parts at nearby MDIs or is highly unlikely due to the large migration energy and an additional energy penalty emerging from the binding energy. The latter also hinders the splitting in the perpendicular direction to the interface plane. As a conclusion, it could be argued that the vacancy clusters of up to four atoms would be trapped near the MDI sites, but in order to prove that further, more complicated NEB calculations would have to be made. In order to estimate the kinetics of vacancy clusters various possible migration paths must be considered. Due to the complex structure of the Cu–Nb interface it is likely that the migration paths are non-trivial and the probabilities of different mechanisms themselves have temperature dependence identifying which from DFT calculations is computationally prohibitively demanding.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. The migration barriers along a straight path from an MDI to the third-nearest MDI. The triangles correspond to the exact calculations, the solid line is a spline interpolation between the exact points and the dashed line is a spline interpolation between the local minima representing the increase in barriers due to the internal strain field. The energies are referenced to vacancy formation energy at an MDI.
Download figure:
Standard image High-resolution image{kind=link}
In conclusion, the vacancy clusters spread over an area which grows with each additional vacancy created. This results in a larger defect capture radius probably increasing the interstitial sink efficiency of the interface. The formation and binding energies show that the interface will be preloaded with vacancy clusters at temperatures above 0 K due to the extremely low formation energy and positive binding energies. Furthermore, the clusters would mainly consist of two and three vacancies. Under irradiation, excess population of vacancies will have a strong driving force towards the interface. Irradiation created interstitials would diffuse fast into the interface and eventually annihilate at the vacancy clusters, providing a mechanisms to increase recombination.
4. Conclusion
In summary, vacancy clusters in the semicoherent Cu–Nb interface were studied using DFT calculations and compared to predictions obtained from EAM potentials. Two main results emerge form this study, first that the predicted vacancy structure is compact, differing notoriously with published work based on EIP, and second that the vacancy clusters containing up to four vacancies have a smaller formation energy than monovacancy in a bulk. Additionally, the binding energies show that the vacancy clusters are energetically stable with an upper limit for the cluster having four vacancies. These last results are qualitatively similar to EIP predictions, differing mostly in the numerical values. The NEB calculations show that the migration of a vacancy from one MDI to another is highly improbable due to the high barriers. These findings suggest that at nonzero temperatures the interface will be preloaded with vacancy clusters with a relatively large capture radius for interstitials in the interface plane. Therefore, the semicoherent Cu–Nb interface seems to be a highly effective sink for point defects that form due to radiation.
Acknowledgments
The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/20072013) under grant agreement no 263273 and Swedish Centre for Nuclear Technology (SKC). Additionally, AT received funding from the Carl Trygger Foundation. Computational resources were provided by Swedish National Infrastructure for Computing (SNIC).