
Abstract
Disorder effects in alloys are usually modeled by averaging various supercell calculations considering different positions of the alloy atoms. This approach, however, is only possible as long as the portion of the individual components of the alloy is sufficiently large. Herein, we present an ab initio study considering the lithium insertion material Li1−x[Ni0.33Co0.33Mn0.33]O2 as model system to demonstrate the power of the coherent potential approximation within the Korringa–Kohn–Rostoker Green's function method. This approach enables the description of disorder effects within alloy systems of any composition. It is applied in this study to describe the (de-)intercalation of arbitrary amounts of lithium from the cathode active material. Moreover, we highlight that using either fully optimized structures or experimental lattice parameters and atomic positions both lead to comparable results. Our findings suggest that this approach is also suitable for modeling the electronic structure of state-of-the-art materials such as high-nickel alloys.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Rechargeable lithium-ion batteries (LiB) are crucial components of portable entertainment, computing and telecommunication devices. In the light of climate change and diminishing fossil-fuel reservoirs their role is becoming increasingly important, e.g.in electric mobility and large-scale energy storage. This leads to an increased research on clean and efficient energy storage materials with the objective of identifying alternative cathode active materials (CAM) with high specific capacity that are cheap, safe, non-toxic, and long-lived.
The first commercially used CAM within LiB was LiCoO2. It was first reported by Goodenough in 1980 [1], and has been studied extensively since [2–4]. The maximum attainable capacity of this material system is found to be around 160 mAh g−1 [5–7]. Drawbacks of LiCoO2 are its poor rate capability and low specific capacity as well as the high cost of cobalt and its toxic character [6, 8]. The replacement of cobalt by nickel leads to an improved specific capacity of around 220 mAh g−1 for LiNiO2 [7, 9, 10]. However, material systems including nickel show unstable cycling performance and capacity fading especially when the material system is charged and discharged at high current rates. The usage of manganese as substitute for cobalt leads to structural issues. Although layered LiMnO2 was found to give a relatively high specific capacity of approximately 200 mAh g−1 and an increased temperature stability, its drawbacks include capacity fading and an electrochemically undesirable structural conversion to the spinel structure during cycling [11, 12].
Currently, alloys with a combination of nickel, cobalt and manganese Li1−x [Nia Cob Mnc ]O2 (NCM) are under investigation, with the hopes of combining the advantages of the individual crystalline components. NCM 622 (the numbers indicate the weighting of the individual components) and materials with higher nickel content are increasingly becoming state-of-the-art technology. This approach, however, is accompanied by the introduction of disorder within CAMs as the transition metal atoms will not be systematically arranged. The associated changes in its electronic band structure affect the overall properties, e.g.of the battery in which they are used. Therefore, it is beneficial to better understand the interplay between electronic band structure, material composition, and the effects of disorder in the system.
The experimental investigation of it, however, is not straightforward, since often only effective (averaged) material parameters can be measured, i.e. the effect of individual alloy constituents cannot be resolved. This is where simulations can bridge a gap and allow for a systematic investigation. The first ab initio calculations for the related high energy and voltage CAM Li2CoMn3O8 were performed by Eglitis and Borstel [13, 14]. The disorder effects in the electronic band structure of alloys are usually modeled using a supercell approach, e.g. with the Vienna ab initio simulation package (VASP). This involves averaging various supercell simulations that consider different positions of alloy atoms in the system consisting of a small number of unit cells. However, disorder within material systems combined with partial (de-)intercalation of lithium makes the use of supercells increasingly impractical.
Thus, it is the purpose of this paper to show the applicability of the Korringa–Kohn–Rostoker (KKR) method [15]. We discuss the effects resulting from disorder within alloy systems by making use of the single-site coherent potential approximation (CPA) within the KKR framework. This approach enables us to describe alloys of any composition. Recent studies in the literature show that it can be successful used to describe, e.g. Fe-based transition metal alloys or semiconducting alloys [16–21]. In this study, we will apply the combinatorial approach of KKR method and CPA to investigate CAMs. We demonstrate that the usage of experimental structure data instead of fully relaxed structures with respect to internal and external cell parameters leads to comparable results. We use the alloy NCM 111 as model system since it is experimentally and theoretically well studied.
2. Computational details
The crystalline CAMs LiNiO2, LiCoO2 and LiMnO2 as well as the alloy NCM 111 are considered as model systems in order to evaluate the comparability between the KKR results and the supercell approach using VASP. Additionally, the influence of employing either experimental structure data or fully relaxed structures with respect to internal and external cell parameters is investigated.
2.1. Structural information of the systems under consideration
Undistorted layered transition metal oxides LiNiO2, LiCoO2, LiMnO2 and NCM 111 crystallize in a hexagonal respectively rhombohedral structure as shown in figure 1. Here, lithium is occupying the 3b site, the transition metal(s) is located at the 3a site, and oxygen is located at the 6c site of the hexagonal unit cell. It can be considered as an α-NaFeO2-type structure with a space-group symmetry of R m which is similar to an ordered rock-salt type where lithium and metal ions occupy alternate (1 1 1) layers. Armstrong et al, however, demonstrated that due to the strong Jahn–Teller nature of the high-spin Mn3+ ion within LiMnO2, the local site symmetry around the manganese ions is distorted from a regular octahedron. In consequence, the crystal structure is not rhombohedral but monoclinic showing a C2/m space group [11, 22, 23]. Within all these structures, the transition metal (TM) ions occupy the octahedral sites in the sublattice of oxygen. Based on crystal field theory, their 3d-bands split into t2g-like and eg-like bands [24–26].
m which is similar to an ordered rock-salt type where lithium and metal ions occupy alternate (1 1 1) layers. Armstrong et al, however, demonstrated that due to the strong Jahn–Teller nature of the high-spin Mn3+ ion within LiMnO2, the local site symmetry around the manganese ions is distorted from a regular octahedron. In consequence, the crystal structure is not rhombohedral but monoclinic showing a C2/m space group [11, 22, 23]. Within all these structures, the transition metal (TM) ions occupy the octahedral sites in the sublattice of oxygen. Based on crystal field theory, their 3d-bands split into t2g-like and eg-like bands [24–26].
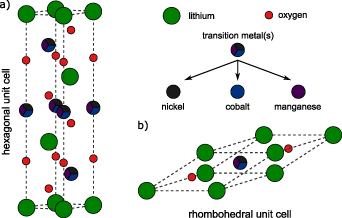
Figure 1. Hexagonal (a) and rhombohedral (b) unit cell of LiMeO2 with Me = [Ni, Co, NCM 111]. It is composed of three respectively one formula unit.
Download figure:
Standard image High-resolution image2.2. Technical description of the modelling process
The fully relativistic version [27] of the screened KKR method [28, 29] has been applied for all computations shown in this study. Experimental structure information published in literature (see table 1) has been used as input. No optimization of the lattice constants and internal atomic positions of fully relaxed structures with respect to internal and external cell parameters have been performed. The hexagonal unit cell was transformed with respect to symmetry issues into a rhombohedral unit cell containing only one formula unit in order to save computational time (see figure 1). The single-site CPA [30, 31] implementation in the KKR framework [32, 33] has been applied for the analysis of equimolar alloying among the crystalline materials LiCoO2, LiNiO2 and LiMnO2. The local spin density approximation (LSDA) [34, 35] has been used for treating the exchange-correlation energy functional. Brillouin zone integration in reciprocal space is done with a 40 × 40 × 40 mesh. Possible ferromagnetic and anti-ferromagnetic spin ordering were used as starting points for self-consistent calculations on NCM 111. Convergence studies were performed with respect to the number of k-points as well as the maximum angular momentum. A broadening of 25 meV was used for the energy points on the contour.
Table 1. Overview of experimental structure parameters used in the computations. Since Jahn–Teller distortion occurs in LiMnO2, it requires a monoclinic instead of hexagonal unit cell.
| Compound | a (Å) | b (Å) | c (Å) | Li-O (Å) | TM-O (Å) |
|---|---|---|---|---|---|
| LiNiO2 [36] | 2.8755 | 2.8755 | 14.1806 | 2.1100 | 1.9700 |
| LiCoO2 [37] | 2.8150 | 2.8150 | 14.0516 | 2.0910 | 1.9221 |
| LiMnO2 [11] | 5.4388 | 2.8086 | 5.3878 | 4 × 2.0970 | 4 × 1.9212 |
| 2 × 2.2897 | 2 × 2.3078 | ||||
| Li[Ni0.33Co0.33Mn0.33]O2 [38] | 2.8602 | 2.8602 | 14.2278 | 2.1048 | 1.9615 |
2.3. Consideration of chemical disorder in alloy systems
The presence of chemical disorder breaks the translational symmetry of the crystal even if a perfect underlying geometrical lattice is assumed. In most computational codes this situation is handled by making use of either the virtual crystal approximation [39–41] or large supercells averaging over several configurations. While the former approach often fails to capture the relevant physics [42–44], the latter approach leads to high computational costs and possible supercell effects [45, 46]. In addition, computing supercells demands excessive computational power considering material systems that contain only a small amount of one component of an alloy system. An alternative approach is the usage of the CPA [30]. Here, the alloy is replaced by an effective medium that approximates its properties but has the periodicity of the underlying lattice. Constructing a configurational average is most simple if any correlation between the occupations of neighboring sites is ignored (single-site CPA). Thus, a random distribution of the components is assumed with the alloy composition being the only restriction. In consequence, short-range order is not considered. The single-site CPA can be extended to include several sites (non-local CPA) [47], taking the effects of correlations due to local clusters in the alloy into account.
The study presented here is based entirely on experimentally observed structure data. As shown by Yin et al for the alloy system NCM 111 using X-ray/neutron diffraction, only one (average) bond length between the TMs (nickel, cobalt, manganese) and oxygen ions could be identified [38]. Hence, the usage of the single-site CPA seems to be reasonable as this method assumes that the bond length between the TM ions and oxygen ions is restricted to one value, even though NCM 111 contains three different TMs.
3. Results and discussion
3.1. Comparison of different crystalline CAMs
The individual crystalline components of the alloy system NCM were examined in order to demonstrate the applicability of the KKR method considering exclusively experimental structure data. Figure 2 depicts spin-resolved density of state (DOS) computations of rhombohedral LiNiO2 and LiCoO2 as well as monoclinic LiMnO2. By evaluating the occupation of the d-bands of the TMs, i.e. the t2g- and eg-band, their oxidation states were determined.

Figure 2. Spin-resolved DOS of rhombohedral LiNiO2 (top), LiCoO2 (middle) and monoclinic LiMnO2 (bottom). Assuming octahedral crystal field splitting, possible spin configurations of the d-states of the TMs are sketched near the upper right corner of each graph. The colored area marks the DOS of the single TMs. Light and dark colors indicate unoccupied/occupied states, respectively.
Download figure:
Standard image High-resolution imageThe computational result considering LiNiO2 (figure 2, top) shows that the Fermi energy comes to lie in the middle of the spin-up eg-band of nickel which is half occupied (mNi = 0.65 μB). Adding in the fully occupied t2g-bands, a low-spin configuration of the d-states like (t2g)6(eg)1 is observed. As a consequence of the empty nickel 4s bands, the oxidation state of nickel is suggested to be +III. This result is in accordance with VASP computations by Anisimov et al and other groups published in the literature [48–50].
Cobalt is one of the few elements that is well known to be ferromagnetic in its pristine form. The DOS of LiCoO2 (figure 2, middle), however, reveals that adding lithium and oxygen to cobalt leads to a non-magnetic spin ordering (mCo = 0.004 μB). Neglecting the fact that the Fermi energy cuts minimally into the upper edges of the t2g-bands, a low-spin configuration of the d-states like (t2g)6(eg)0 is indicated. Since the 4s bands are unoccupied, the oxidation state of cobalt is suggested to be +III. Calculating the band gap between the t2g- and eg-bands gives a value of 0.77 eV which is underestimated by a factor of approximately three. Experimentally, the material is classified as a semiconductor with a band gap in the range of 2.1–2.7 eV [51]. The deviation, however, is not related to the KKR method. Our result, i.e. the position of the Fermi energy and the shape of the DOS, is in agreement with ab initio computations by Koyama et al and other groups which considered fully relaxed structures and used VASP to perform the simulations [26, 52–55].
While pristine manganese is not ferromagnetic, it acquires a magnetic moment when combined with lithium and oxygen to form LiMnO2. The calculated DOS (figure 2, bottom) highlights that the material is strongly ferromagnetic (mMn = 3.50 μB). Only the t2g- and half of the eg-band of the manganese spin up channel are occupied. This indicates a high-spin configuration of the d-states (t2g)3(eg)1 and an oxidation state of +III of manganese. Our computational result is comparable to the simulations by Huang et al and other groups shown in the literature [26, 52, 56, 57].
At this state, we can already conclude that the results obtained by means of VASP and the KKR method agree well regarding crystalline systems. We demonstrated that the electronic structure of these crystalline CAMs can also be obtained when using exclusively experimental input parameters, i.e. lattice constants and internal atomic positions. The excellent agreement indicates not only a very good quality of the experimental data but also highlights the strong link between experimental results and theoretical simulations in general.
3.2. Consideration of alloying between the crystalline systems
Current research efforts for commercial applications are focusing on complex alloy systems instead of crystalline materials. Here, the proportion of the individual components can vary strongly between single-digit and high double-digit percentages. Describing such systems by means of large supercells is challenging and not always possible. The standard KKR method can be extended for describing alloys by using the CPA to consider systems with arbitrary proportion of alloy atoms. NCM 111 is well suited as a model system to evaluate the CPA approach since it has been extensively studied experimentally and theoretically. We want to note, that, first, we compare the supercell approach within VASP with our CPA-KKR approach and, secondly, we assess the quality of results obtained with fully optimized or experimentally obtained structural data.
The upper part of figure 3 depicts the spin-resolved DOS of fully lithiated NCM 111 (see discussion in section 3.4 on delithiated NCM 111). The comparison with the DOS of the crystalline computations (see figure 2) reveals, that the shape of the NCM 111 DOS is broadened and smoothened. This effect is directly related to the disorder of the three transition metals in the system. Based on the KKR computation, NCM 111 can be classified as a half-metal. The material has a band gap of about 0.4 eV for the spin down channel and the position of the Fermi energy is located within a spin-up band. The individual TMs in the system differ in their magnetic properties. Cobalt exhibits a diamagnetic behavior, and in principle, its d-bands and the position of the Fermi level are similar to the LiCoO2 computation indicating the presence of low-spin Co3+ ions. Contrary to this, nickel is ferromagnetic in accordance to the LiNiO2 calculation. The comparison between the DOS of NCM 111 and LiNiO2 reveals a slight shift of the Fermi energy towards the upper edge of the nickel eg-band. Hence, in NCM 111 more nickel states are occupied indicating the presence of high-spin Ni2+ as well as low-spin Ni3+ ions. The character of the manganese ions changes even more dramatically between NCM 111 and monoclinic LiMnO2. Both manganese eg-bands are empty whereas the whole t2g-band of the spin down channel as well as part of the t2g-band of the spin up channel are occupied. Consequently, low-spin Mn3+ ions as well as high-spin Mn4+ ions might be present. The conclusions highlight that an anti-ferromagnetic coupling of the magnetic moments of nickel and manganese is energetically favorable for the system.

Figure 3. Spin-resolved DOS computed for NCM 111 at different state-of-charges. The colored area marks the DOS of the single TMs. Light and dark colors indicate unoccupied/occupied states, respectively.
Download figure:
Standard image High-resolution imageOur result differs somewhat from supercell calculations by Koyama et al and Hwang et al using VASP and fully relaxed structures [24–26, 52]. Specifically, the position of the nickel eg- and manganese t2g-band of the spin-up channel relative to the Fermi energy is slightly different. While our computations predict a partial filling of both bands, the VASP calculations published in the literature suggest that these bands are fully occupied and completely empty, respectively. Therefore, there is no evidence for the presence of Mn3+ and Ni3+ ions within those simulations. Comparing the computed results to X-ray photoemission spectroscopy (XPS) and X-ray absorption spectroscopy (XAS) measurements that are shown in literature [58–60], the presence of trivalent nickel and manganese ions within fully lithiated NCM 111 cannot be ruled out [61]. Anyway, it is experimentally confirmed that the predominant species within the material system are Ni2+, Co3+ and Mn4+. In this particular case, the VASP calculations seem to provide a better description of the experimental behavior.
However, smaller deviations regarding both approaches for describing alloys, i.e. the supercell calculation using fully optimized structures compared to the single-site CPA approach considering experimental structure data, were to be expected. While in the VASP calculations different bond lengths between the individual TMs and oxygen atoms were assumed, experimentally only one (average) bond length could be observed. Thus, we also neglect short-range order effects, i.e. clustering and a possible Jahn–Teller distortion of any TM sites. The consideration of short-range order effects, however, might be crucial for explaining the different observations. Although both approaches differ in the subtleties, we want to emphasize that the qualitative changes in the DOS going from the crystalline systems to the alloy are the same, i.e. the DOS of cobalt is comparable to the crystalline system while the occupation of the nickel and manganese states increases respectively decreases indicating different oxidation states compared to the crystalline systems.
3.3. Consideration of disorder effects due to cation mixing
It is well known that the usage of nickel within CAMs leads to the occurrence of cation mixing between nickel and lithium ions during the synthesis [62–66]. Regarding LiNiO2, this is a direct consequence of the transformation of unstable Ni3+ ions into more stable Ni2+ ions, which then move to the lithium layer. The cation mixing is based on the similar size of Ni2+ (0.69 Å) and Li+ (0.72 Å) ions [67, 68]. Due to the small amount of structural disorder (typically less than 10%), the effect cannot be efficiently modeled using supercells. With the CPA approach, however, this small amount of disorder can be easily translated into changes of the electronic structure of the material.
The upper part of figure 4 shows the DOS of LiNiO2 considering 5% of site-exchange between nickel and lithium. Except for a smooth broadening, the DOS of nickel ions at the 3b site stays relatively unchanged. Although the 3a crystallographic site is octahedrally coordinated as well, the eg- and t2g-bands of nickel can barely be identified. However, the computation indicates that the magnetic moments of nickel atoms at the different crystallographic sites couple anti-ferromagnetically (mNi(3b),95% = 0.51 μB, mNi(3a),5% = −0.23 μB). Moreover, the band gap of the spin down channel is reduced as a consequence of the site exchange.

Figure 4. Spin-resolved DOS of disordered LiNiO2 (top) and NCM 111 (bottom) considering a site exchange of 5% and 3%, respectively. The colored area marks the DOS of nickel. Light and dark colors indicate unoccupied/occupied states, respectively.
Download figure:
Standard image High-resolution imageThe same effect occurs considering NCM 111. It is experimentally found that cation mixing affects less than 5% of the sites. The lower part of figure 4 depicts the spin-resolved DOS with 3% site exchange disorder of nickel atoms at the 3a crystallographic site. The separation of the nickel eg- and the t2g-bands seems to have vanished at the 3a site. The majority of the d-bands of both spin states is located below the Fermi energy. In accordance with the computation on LiNiO2, the nickel atoms at different crystallographic sites in the structure couple anti-ferromagnetically (mNi,3a = −0.47 μB, mNi,3b = 0.77 μB). Moreover, the band gap of the spin down configuration is reduced due to the cationic mixing.
3.4. Modelling of the electronic structure of NCM 111 at different state-of-charges
Lithium is continuously (de-)intercalated from the CAM during the life cycle of an electrochemical device. This process is accompanied by changes in the electronic structure of the material. Using supercells is only partially suitable to study these transformations in detail since it is restricted to discrete values for the lithium content within the structure. In contrast, the KKR framework together with the CPA can be used to describe the electronic structure of CAMs with an arbitrary amount of lithium within the unit cell. As a model system, the electronic structure of NCM 111 is considered at different state-of-charges. We assume that all structural data, i.e. lattice constant and internal atomic positions, remain unchanged compared to the fully lithiated case. Based on experimental findings by Yin et al using X-ray/neutron diffraction, our assumption is reasonable since the lattice parameters, the interatomic distance between the lithium and oxygen atoms, and also the bond length between the transition metals and the oxygen atoms changed by only around 2% during the delithiation process of Li1−x [Ni0.33Co0.33Mn0.33]O2 [38]. This result is also in accordance to XAS measurements, performed by Yoon et al [58].
Figure 3 shows the spin-resolved DOS of NCM 111 calculated for different amounts of lithium (x = 0, 1/3, 2/3, 1) within the structure. Considering the magnetic behavior of the TMs, our computations indicate that nickel and manganese couple antiferromagnetically in the range between 0 ⩽ x ⩽ 0.55 whereas cobalt shows no magnetic moment. With further delithiation of the material, the magnetic moment of nickel vanishes. In contrast, cobalt forms a magnetic moment that couples ferromagnetically with the magnetic moment of manganese. The theoretical results published in the literature on this subject using supercell approaches are contradictory. While VASP computations performed by Hwang et al suggest that anti-ferromagnetic coupling between cobalt and manganese is energetically favorable [24], Koyama et al assumed ferromagnetic spin ordering [25, 26, 52]. The consideration of the total energy of both spin configurations in our computations, however, revealed that the ferromagnetic coupling is energetically more favorable.
Similar to the fully lithiated case, we attempted to identify the oxidation state of the TMs at different state-of-charges. It seems that redox reactions take place in distinct regions of delithiation space indicated by changing magnetic moments of the TMs as a function of the lithium content within the structure (see figure 5). The magnetic behavior of nickel and manganese changes simultaneously in the interval 0 < x ⩽ 0.55, while it stays constant for cobalt. This range is in agreement with XANES measurements performed by Yoon et al who showed that the Ni edge shifts until 51% state-of-charge substantially to higher energies [58, 59]. Since the magnetic moment of manganese changes only slightly in contrast to nickel, the following oxidation reactions are assumed: Ni2+/Ni3+ for 0 < x ⩽ 0.2 and Ni3+/Ni4+ for 0.2 < x ⩽ 0.55. Due to the fact that most manganese ions in fully lithiated NCM 111 are already in the oxidation state +IV, the oxidation reaction Mn3+/Mn4+ for 0 < x ⩽ 0.55 is not that pronounced. For lithium contents lower than 45%, cobalt is the dominant electrochemical active species. Hence, the cobalt ions are oxidized Co3+/Co4+ in the range between 0.55 < x ⩽ 1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. Fundamental changes in the magnetic structure of the TMs during the delithiation process. At around x = 0.55, a change of the electrochemically active species from nickel to cobalt occurs. The highlighted regions (yellow) underline the state-of-charge (0, 1/3, 2/3 and 1) that are accessible through a supercell approach considering a single unit cell.
Download figure:
Standard image High-resolution image{kind=link}
As discussed in the context of the fully lithiated NCM 111, slight deviations in the DOS occur due to the usage of experimental structure data and thus the neglect of short-range ordering compared to optimized structure data. Although these deviations can also be found here, the qualitative behavior regarding oxidation states agrees well with supercell calculations considering four distinct state-of-charge published in the literature [24–26, 52]. The results, however, are only meant to be a guidance to show the connection between the lithium content within the structure and the electrochemical activity of the TMs since structural changes are not considered in our computations. Nonetheless, our results show impressively that the entire loading range can be modeled and that the simulations provide significant additional insights.
4. Conclusions
In summary, density functional theory calculations on several layered CAMs were performed using the KKR method exclusively considering experimentally determined structure information. We demonstrated that the computational results for crystalline LiNiO2, LiCoO2 and LiMnO2 agree well with simulations using VASP published in the literature considering fully relaxed structures with respect to internal and external cell parameters. Nowadays, alloys are frequently used as CAMs. We used the single-site CPA within the KKR framework to adequately describe the disorder in such systems. For this purpose, NCM 111, as a material that has been extensively studied theoretically and experimentally, was considered as a model system. Although our calculations are in good qualitative agreement with VASP simulations published in the literature, it seems that experimental structural data is not sufficient to account for subtleties in the electronic structure of such a complex system. The missing short-range order, which could not be resolved experimentally but theoretically in fully relaxed structures, was identified as a possible cause for the deviations.
The results using the CPA approach to describe disorder in alloys, however, ar nonetheless promising. To demonstrate the wide range of possible applications, the influence of small amounts (less than 5%) of site-exchange between Li+/Ni2+ ions on the DOS of LiNiO2 and NCM 111 was investigated. In addition, changes in the electronic structure when extracting an arbitrary amount of lithium from the CAM were simulated using NCM 111 as a model system. The change in magnetization of the three TMs as a function of the lithium content within the structure gives a good guidance when the oxidation processes of the individual transition metals take place. Such modeling is challenging when using supercells due to restrictions regarding the size of the system under consideration. However, it is worth noting that the CPA within the KKR framework can also be applied to optimized structures making the description of subtleties in the electronic structure of any kind of alloy possible. Thus, it provides a powerful tool to describe state-of-the-art materials, e.g. CAMs with high nickel content and other alloying elements in the low percentage range.
Acknowledgments
We appreciate helpful discussions with Sebastian Benz about XPS results and the oxidation states of the TM atoms. We acknowledge computational resources provided by the HPC Core Facility and the HRZ of the Justus-Liebig-University Giessen. We would like to thank Dr Marcel Giar of HPC-Hessen, funded by the State Ministry of Higher Education, Research, and the Arts, for programming advice. Financial support is provided by the DFG via the GRK (Research Training Group) 2204 'Substitute Materials for Sustainable Energy Technologies'.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).