
Abstract
With the recent discovery of in-plane chemically ordered MAX phases (i-MAX) of the general formula ( )2AC comes addition of non-traditional MAX phase elements. In the present study, we use density functional theory calculations to investigate the electronic structure, bonding nature, and mechanical properties of the novel (W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC i-MAX phases. From analysis of the electronic structure and projected crystal orbital Hamilton populations, we show that the metallic i-MAX phases have significant hybridization between W and C, as well as Sc(Y) and C states, indicative of strong covalent bonding. Substitution of Sc for Y (M2) leads to reduced bonding strength for W–C and Al–Al interactions while M2–C and M2–Al interactions are strengthened. We also compare the Voigt–Reuss–Hill bulk, shear, and Young's moduli along the series of M1 = Cr, Mo, and W, and relate these trends to the bonding interactions. Furthermore, we find overall larger moduli for Sc-based i-MAX phases.
)2AC comes addition of non-traditional MAX phase elements. In the present study, we use density functional theory calculations to investigate the electronic structure, bonding nature, and mechanical properties of the novel (W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC i-MAX phases. From analysis of the electronic structure and projected crystal orbital Hamilton populations, we show that the metallic i-MAX phases have significant hybridization between W and C, as well as Sc(Y) and C states, indicative of strong covalent bonding. Substitution of Sc for Y (M2) leads to reduced bonding strength for W–C and Al–Al interactions while M2–C and M2–Al interactions are strengthened. We also compare the Voigt–Reuss–Hill bulk, shear, and Young's moduli along the series of M1 = Cr, Mo, and W, and relate these trends to the bonding interactions. Furthermore, we find overall larger moduli for Sc-based i-MAX phases.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Mn+1AXn (MAX) phases, where M is an early transition metal, A is an A-group element (mostly fro m group 13 and 14) and X is C and/or N, are a family of atomically layered ceramics that consist of Mn+1Xn sheets sandwiched in between one atom thick A-layers [1–3]. To date ~70 ternary MAX phases have been synthesized. The large interest in these materials stems from their unique combination of metallic and ceramic attributes [3, 4], with properties such as oxidation resistance and self-healing characteristics [5, 6], reversible deformation [7], and magnetism [8–10]. In recent years, an increased focus has been directed towards using MAX phases as parent material for its two-dimensional (2D) derivative, MXene, realized from selective etching of the A-element [11, 12]. Even though MXenes are a comparatively young family of 2D materials, it has shown high promise for, e.g. energy storage [13] and electromagnetic interference shielding [14].
Adding a fourth element to the MAX phase by alloying on the M-, A- and/or X site allows for even more elemental combinations. Historically, alloying through solid solutions is the primary route investigated [8, 15–21] to, e.g. incorporate magnetic elements or tailoring thermal expansion. However, recent findings demonstrate chemically ordered MAX phases, where the underlying crystal lattice or symmetry governs the stoichiometry. The first example is out-of-plane ordered MAX phases (o-MAX) like Cr2TiAlC2 [22], Mo2TiAlC2 [23, 24], Mo2Ti2AlC3 [24], Mo2ScAlC2 [25], defined by alternating M-layers of two different M elements, where each layer is based on one M-element only. The second example is in-plane ordering (i-MAX), shown for (Mo2/3Sc1/3)2AlC [26, 27], (Mo2/3Y1/3)2AlC [27, 28], (V2/3Zr1/3)2AlC [28], (Cr2/3Sc1/3)2AlC [29], (Cr2/3Y1/3)2AlC [29], and most recently (W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC [30]. The i-MAX phases are defined by a 2:1 ratio of two different M elements with the general formula ( )2AC. Moreover, it has been demonstrated that by detailing the chemical etching of an i-MAX phase, one can obtain either a chemically ordered alloy MXene from selectively etching Al only [31], or a MXene with ordered vacancies by etching Al as well as Sc/Y. Examples of the latter are W1.33C [30] and Mo1.33C [26] MXene.
)2AC. Moreover, it has been demonstrated that by detailing the chemical etching of an i-MAX phase, one can obtain either a chemically ordered alloy MXene from selectively etching Al only [31], or a MXene with ordered vacancies by etching Al as well as Sc/Y. Examples of the latter are W1.33C [30] and Mo1.33C [26] MXene.
In this work, we have used first-principles calculations to investigate the electronic, vibrational, and mechanical properties of (W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC i-MAX phases, motivated by their recent discovery, and being the first W-based MAX phase materials. Both (W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC are stable with a calculated formation enthalpy of −27 and −22 meV/atom, respectively [30]. However, neither of the ternary MAX phases W2AlC, Sc2AlC or Y2AlC have been synthesized, explained by Meshkian et al showing that these are far from being theoretically stable, with a calculated formation enthalpy of +148, +118, and +185 meV/atom, respectively [30]. The finding of i-MAX phases, allowing introduction of non-traditional MAX phase elements like Sc, Y and W, may alter or introduce new properties as compared to previously known MAX phases. This motivates their exploration, for fundamental understanding, and potential future property tailoring.
2. Computational methods
All calculations were performed within the framework of density functional theory as implemented in the Vienna ab initio simulation package [32–34], combined with the Perdew–Burke–Ernzerhof generalized gradient approximation (PBE-GGA) [35] and the projector augmented wave (PAW) method [36, 37]. The unit cells of both compounds were converged to an accuracy of 0.1 meV/atom, using a Monkhorst–Pack [38] 13 × 7 × 5 k-point grid and a plane wave cutoff energy of 400 eV. To calculate the density of states (DOS) and the projected crystal orbital Hamilton population (pCOHP) for each alloy, we used the LOBSTER code [39]. Dynamic stability in terms of phonon dispersion for both i-MAX phases was calculated from 1 × 2 × 1 supercells using the finite displacement method. PHONOPY [40] was used both to create the displacements and for the postprocessing analysis.
Using the energy-strain method [41], we derived and calculated the single crystal elastic constants, where a number of different strains are applied to the crystal lattice, followed by a calculation of the energy associated with each strain. For the monoclinic C2/c structure of (W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC, 14 different strains are needed to obtain the 13 independent elastic constants Cij,. Here we used strain parameters of 0, ±0.01, and ±0.02. From Cij we calculated the bulk modulus (B), shear modulus (S), and Young's modulus (E) within the Voigt (V) and Reuss (R) model as expressed by the equations




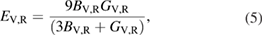
where the Cij's are elastic constants and the Sij's are compliance constants given by an inversion of the elastic-constant matrix. The upper boundary (Voigt) is found assuming that the strain is everywhere uniform while the lower boundary (Reuss) is found assuming that the stress is everywhere uniform. Arithmetic averages of Voigt and Reuss moduli are interpreted as the ratio of average stress and average strain within the composite. The stress and strain are generally unknown in the material and are expected to be nonuniform.
Schematics were produced with VESTA [42].
3. Results and discussion
3.1. Structural properties
(W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC crystallize in a monoclinic C2/c structure [30], like the isostructural (Mo2/3Sc1/3)2AlC [26, 27], (Mo2/3Y1/3)2AlC [27, 28], (V2/3Zr1/3)2AlC [28], (Cr2/3Sc1/3)2AlC [29], and (Cr2/3Y1/3)2AlC [29] i-MAX phases. It should, however, be noted that a closely related orthorhombic Cmcm structure have also been identified for (Mo2/3Y1/3)2AlC, in line with observations for, e.g. the (Cr2/3Y1/3)2AlC i-MAX [29]. The close relation between the C2/c and Cmcm structures have also been shown to be almost degenerate in energy [28, 29, 43].
The monoclinic unit cell of (W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC contains 48 atoms, 12 formula units (Z = 12). Table 1 show the calculated lattice parameters together with atomic positions. Our calculated results are in good agreement with the experimental values with deviations for (W2/3Sc1/3)2AlC lattice parameters a, b, c, and β as −0.45%, −0.13%, +0.06%, and +0.36%, respectively. From table 1 we observe that the exchange of Sc for Y results in increased lattice parameters, as expected with larger metallic radius for Y (1.80 Å) as compared to Sc (1.62 Å).
Table 1. Calculated and experimental structural parameters and atomic positions for (W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC with C2/c symmetry.
| Structural parameters | (W2/3Sc1/3)2AlC | (W2/3Y1/3)2AlC | ||
|---|---|---|---|---|
| Calculated data | Experimental data [30] | Calculated data | Experimental data [30] | |
| a (Å) | 9.326 82 | 9.368 74 | 9.543 59 | 9.515 48 |
| b (Å) | 5.397 68 | 5.404 56 | 5.519 23 | 5.499 32 |
| c (Å) | 13.968 92 | 13.960 75 | 14.127 72 | 14.227 57 |
| α (°) | 90.000 00 | 90.0000 | 90.000 00 | 90.0000 |
| β (°) | 103.458 70 | 103.0908 | 103.623 40 | 103.3302 |
| γ (°) | 90.000 00 | 90.0000 | 90.000 00 | 90.0000 |
| W1 (8f) | (0.772 08, 0.421 65, 0.079 58) | (0.7725, 0.4250, 0.0821) | (0.770 10, 0.423 93, 0.077 05) | (0.2679, 0.3677, 0.0863) |
| W2 (8f) | (0.110 26, 0.405 80, 0.079 80) | (0.1111, 0.4149, 0.0832) | (0.110 00, 0.402 89, 0.077 02) | (0.1113, 0.4664, 0.0724) |
| M2 (8f) | (0.458 23, 0.419 13, 0.110 74) | (0.4529, 0.3878, 0.0907) | (0.461 06, 0.419 47, 0.118 76) | (0.9500, 0.2906, 0.1411) |
| Al1 (8f) | (0.241 17, 0.153 92, 0.251 43) | (0.2373, 0.1236, 0.2535) | (0.247 71, 0.160 01, 0.251 77) | (0.2403, 0.1305, 0.2400) |
| Al2 (4e) | (0.000 00, 0.431 39, 0.250 00) | (0.0000, 0.3506, 0.2500) | (0.000 00, 0.420 14, 0.250 00) | (0.0000, 0.4117, 0.2500) |
| C1 (8f) | (0.916 46, 0.250 25, 0.000 09) | (0.9316, 0.2530, 0.0000) | (0.913 55, 0.259 19, 0.000 07) | (0.9221, 0.2600, 0.0000) |
| C2 (4c) | (0.250 00, 0.250 00, 0.000 00) | (0.2500, 0.2500, 0.0000) | (0.250 00, 0.250 00, 0.000 00) | (0.2500, 0.2500, 0.0000) |
3.2. Electronic structure and bonding analysis
The electronic structure and nature of bonding is essential to explain and understand many physical properties of materials. In addition to calculations of the electronic band structure, see figure 1 and an evident metallic character of both compounds, the bonding characteristics of monoclinic (W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC have been analyzed in terms of the DOS and the pCOHP, as shown in figure 2. In order to facilitate interpretation and to preserve the analogy for crystal orbital overlap population (COOP) analysis, the results are here presented as –pCOHP, rather than pCOHP. Except for the W–W (2) interaction across the Al-layer, see notation in figure 2, only nearest-neighbor interactions are considered in the pCOHP analysis, depicted in figures 2(g) and (h), since these are by far the strongest. Both DOS and pCOHP display five main regions; (i) from −13 to −11 eV, showing localized C-s states interacting mainly with W and Sc or Y, (ii) between −9 and −2 eV, from localized Al-s states in Al–Al interactions, (iii) between −7 and −3.4 eV, with Al-p states in Al–Al interactions as well as the bonding (interacting) states of M-d (W, Sc, Y) and C-p, (iv) from −3.4 to −1 eV, from M-d and Al-p states in bonding Al–Al, W–Al, Sc–Al, Y–Al interactions, (v) and states from −1 eV up to the Fermi level (Ef), mainly dominated by W-d with some contribution from Sc-d or Y-d. The last region shows a significant number of electrons, (N[Ef] = 2.25 states/fu for M2 = Sc and 2.08 states/fu for M2 = Y) despite the low contribution from pCOHP, which mainly consists of W–Sc and W–Y interaction. This is indicative of non-bonding electrons, mainly from the transition metals W and Sc or Y. The non-zero contribution at Ef is also a strong indication of metallic character of these i-MAX phases.
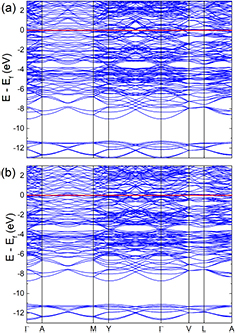
Figure 1. Calculated electronic bandstructure (a) (W2/3Sc1/3)2AlC and (b) (W2/3Y1/3)2AlC.
Download figure:
Standard image High-resolution image
Figure 2. Calculated (a) and (d) electron density of states, (b) and (e) projected crystal orbital Hamilton population (pCOHP), and (c) and (f) integrated projected crystal orbital Hamilton population (IpCOHP) in (a)–(c) (W2/3Sc1/3)2AlC and (d)–(f) (W2/3Sc1/3)2AlC. Schematic along (g) [0 1 0] and (h) [1 1 0] zone axes for considered interactions used in pCOHP analysis.
Download figure:
Standard image High-resolution imageFrom pCOHP we also find anti-bonding interactions below Ef; around −5 to −4 eV by C–C and from −2.4 eV up to Ef by W–C. This is an indication of a non-optimized electronic structure which could be related to an unbalanced number of electrons. Here we have assumed complete occupation of the carbon sites, however, theoretical as well as experimental investigation of C occupancy is the scope of future work. Since the contribution from anti-bonding interactions is rather small, a small amount of, e.g. carbon vacancies could possibly counteract this interaction. In related MAX phase materials, i.e. V4AlC3 [44, 45] and Nb4AlC3 [46], carbon vacancies have been shown theoretically and experimentally to be possible, and to stabilize the materials.
By integrating pCOHP up to Ef, it is possible to get a rough estimate of the relative bond strengths within each i-MAX phase. IpCOHOP in figures 2(c) and (f) suggests the following order, in terms of bond strength for interactions defined in figures 2(g) and (h), for (W2/3Sc1/3)2AlC: C–W > Al–Al > C–Sc ≈ Al–W > W–W (2) ≈ W–Sc > W–W (1) ≈ Al–Sc > C–C. For (W2/3Y1/3)2AlC the corresponding order is C–W > Al–Al > C–Y > Al–W ≈ Al–Y > W–W (2) ≈ W–Y > W–W (1), C–C. The two phases thus differ primarily in that the Al–Sc bonds are significantly weaker relative to most other bonds in (W2/3Sc1/3)2AlC, in contrast to the corresponding Al–Y bonds in (W2/3Y1/3)2AlC. Moreover, similar to regular MAX phases [47], the M–X bonds are stronger than the M–A bonds. On the other hand, the in-plane nearest neighbor bonds are comparatively weak, with the exception for the Al–Al nearest neighbor bonds.
3.3. Phonon dispersion and density of states
The phonon dispersion between high symmetry points in (W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC are shown in figures 3(a) and (c), with only positive frequencies implying their dynamic stability, i.e. stability with respect to lattice vibrations. Their monoclinic crystal structure contains 48 atoms, giving rise to totally 144 phonon branches out of which three are acoustic modes and 141 optical modes. Both i-MAX phases exhibit similar phonon dispersions, which can be related to the similar bonding characteristics discussed in a previous section, with a band gap around 13 THz separating high-frequency contribution, that mainly comes from the light carbon atoms (figures 3(b) and (d)), from the heavier transition metals and Al below the band gap.
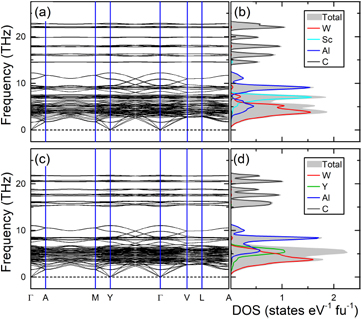
Figure 3. Phonon dispersion (left panels) and total and atomic phonon density of states (right panels) in (a) and (b) (W2/3Sc1/3)2AlC and (c) and (d) (W2/3Y1/3)2AlC.
Download figure:
Standard image High-resolution imageFigures 3(b) and (d) shows the phonon partial density of states (PHDOS) of (W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC where lowest frequency peaks are mainly attributed to W followed by Y(Sc) and Al. The contribution from W is similar for both i-MAX phases. However, upon substitution of Sc for Y the phonon DOS contribution from Al and Y is shifted to lower frequencies. The observed shift for Y, compared to Sc, can be attributed to the fact that it is heavier than Sc. The shift for Al can be attributed to the stronger Al–Y bonds as compared to Al–Sc, seen in figure 2.
3.4. Mechanical and elastic properties
The mechanical and elastic properties are dependent on the crystal structure and bonding between atoms. In table 2 we present calculated single crystal elastic constants Cij at 0 GPa for (W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC along with polycrystalline Voigt moduli; bulk modulus B, shear modulus G, and Young's modulus E as derived from Cij using equations (1), (3) and (5). In addition, we also present the Reuss moduli, calculated from the compliance constants obtained by inverting the 6 × 6 elastic-constant matrix, derived using equations (2), (4) and (5), together with the arithmetic means of the Voigt and Reuss moduli, i.e. the Voigt–Reuss–Hill (VRH) averages. Both i-MAX phases investigated herein fulfill the mechanical stability criterion for monoclinic structures [48].
Table 2. Calculated elastic constants Cij and Voigt (V), Reuss (R) and VRH moduli (in GPa) of (W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC. The Reuss moduli have been calculated from the compliance constants obtained by inverting the 6 × 6 elastic-constant matrix.
| (W2/3Sc1/3)2AlC | (W2/3Y1/3)2AlC | |
|---|---|---|
 |
319 | 293 |
 |
117 | 114 |
 |
117 | 111 |
 |
85 | 89 |
 |
301 | 295 |
 |
85 | 80 |
 |
65 | 74 |
 |
318 | 309 |
 |
78 | 86 |
 |
112 | 110 |
 |
−0.74 | −1.26 |
 |
91 | 92 |
 |
92 | 94 |
 |
175 | 167 |
 |
112 | 92 |
 |
143 | 130 |
 |
100 | 99 |
 |
88 | 83 |
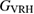 |
94 | 91 |
 |
253 | 247 |
 |
209 | 192 |
 |
231 | 219 |
First to note is that substitution of Sc for Y in general leads to decreased moduli. This decrease is greater for the Reuss moduli than for the Voigt moduli. We also note that the Reuss moduli are significantly lower than the Voigt moduli, in particular when it comes to the bulk modulus:  is 37% lower than
is 37% lower than  for (W2/3Sc1/3)2AlC, and 45% lower than
for (W2/3Sc1/3)2AlC, and 45% lower than  for (W2/3Y1/3)2AlC. A similarly large difference is seen for the two i-MAX phases (Mo2/3Sc1/3)2AlC and (Mo2/3Y1/3)2AlC, which suggests that it is important to calculate both Voigt and Reuss moduli for i-MAX phases [27].
for (W2/3Y1/3)2AlC. A similarly large difference is seen for the two i-MAX phases (Mo2/3Sc1/3)2AlC and (Mo2/3Y1/3)2AlC, which suggests that it is important to calculate both Voigt and Reuss moduli for i-MAX phases [27].
Comparison of calculated moduli of (W2/3Sc1/3)2AlC with the hypothetical W2AlC (BV = 214 GPa, GV = 110 GPa, EV = 280 GPa) and Sc2AlC (BV = 88 GPa, GV = 57 GPa, EV = 140 GPa) in [49] shows that all moduli are in-between the two M2AX phases. Both GV and EV are strengthened by ~10% as compared to the arithmetic mean for the two M2AX phases. Generally, a large shear modulus G of a material is an indication of well-defined directional bonding between atoms. Here, GV is 100 and 99 GPa for (W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC, respectively. This indicates rather similar bonding, or more likely, a comparable total bond strength in the two i-MAX phases, which is confirmed by the bonding analysis in figure 2 where W–C and Al–Al is found to be slightly stronger for (W2/3Sc1/3)2AlC compared to (W2/3Y1/3)2AlC. This is, however, compensated by the latter having stronger interactions between C–Y and Al–Y as compared to C–Sc and Al–Sc.
3.5. Comparison of i-MAX phases where M1 = Cr, Mo, and W
To investigate the effect of M-element on bonding and related properties in i-MAX, we depict IpCOHP for five selected interactions and the VRH moduli as function of M1 in ( )2AlC and (
)2AlC and ( )2AlC for M1 = Cr, Mo, and W. VRH values for Cr- and Mo-based i-MAX phases are from [27, 29] while IpCOHP values for Mo-based i-MAX phases are taken from [27]. All interactions, except for Al–M2 and C–M2, are stronger when M2 = Sc. For both Sc and Y, we find that both C–M and Al–M interactions are strengthened when going from Cr to Mo to W. For the C–M2, Al–Al, and Al–M2 there is a decrease when going from Cr to Mo followed by no change or a slight increase in their interaction when going from Mo to W. These trends in interaction strengths are reflected in the trends found for the moduli in figure 4(b). The moduli for Cr-based i-MAX are in most cases higher than for Mo-based ones, which can be related to the comparatively stronger Al–Al and C–M2 interactions. The increased moduli when going from Mo to W can be explained by the steady decrease in Al–M interaction strength and the dominating C–M interaction while the other interactions are unaffected. These non-linear trends found for moduli and for some of the bonding interactions for Cr, Mo, and W may be a result from size differences for M1 and M2 as well as different electronegativities. The size of the M1 atom (1.28 Å for Cr and 1.39 Å for Mo and W) may explain why selected bonding interactions are changed from Cr to Mo but not from Mo to W. This is most notable for the Al–Al interaction. Moreover, the electronic properties will also influence the bonding interactions where both Mo (2.16) and W (2.36) are significantly more electronegative than Cr (1.66). Finally, comparing the moduli with a well-known traditional MAX phase, Ti2AlC with B = 138 GPa, G = 113 GPa, E = 267 GPa [50], we find similar B but lower G and E for the i-MAX phases.
)2AlC for M1 = Cr, Mo, and W. VRH values for Cr- and Mo-based i-MAX phases are from [27, 29] while IpCOHP values for Mo-based i-MAX phases are taken from [27]. All interactions, except for Al–M2 and C–M2, are stronger when M2 = Sc. For both Sc and Y, we find that both C–M and Al–M interactions are strengthened when going from Cr to Mo to W. For the C–M2, Al–Al, and Al–M2 there is a decrease when going from Cr to Mo followed by no change or a slight increase in their interaction when going from Mo to W. These trends in interaction strengths are reflected in the trends found for the moduli in figure 4(b). The moduli for Cr-based i-MAX are in most cases higher than for Mo-based ones, which can be related to the comparatively stronger Al–Al and C–M2 interactions. The increased moduli when going from Mo to W can be explained by the steady decrease in Al–M interaction strength and the dominating C–M interaction while the other interactions are unaffected. These non-linear trends found for moduli and for some of the bonding interactions for Cr, Mo, and W may be a result from size differences for M1 and M2 as well as different electronegativities. The size of the M1 atom (1.28 Å for Cr and 1.39 Å for Mo and W) may explain why selected bonding interactions are changed from Cr to Mo but not from Mo to W. This is most notable for the Al–Al interaction. Moreover, the electronic properties will also influence the bonding interactions where both Mo (2.16) and W (2.36) are significantly more electronegative than Cr (1.66). Finally, comparing the moduli with a well-known traditional MAX phase, Ti2AlC with B = 138 GPa, G = 113 GPa, E = 267 GPa [50], we find similar B but lower G and E for the i-MAX phases.

{kind=link}
{kind=link}
{kind=link}
Figure 4. (a) IpCOHP for five selected interactions and (b) Voigt–Reuss–Hill (VRH) moduli as function of M in (M2/3Sc1/3)2AlC and (M2/3Y1/3)2AlC where M = Cr, Mo, and W [27, 29].
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusions
Two new atomically laminated W-based compounds, (W2/3Sc1/3)2AlC and (W2/3Y1/3)2AlC, belonging the family of i-MAX phases, have recently been synthesized, and are herein theoretically explored in terms of electronic structure, bonding characteristics, and mechanical properties. Both phases are described by a monoclinic structure of space group C2/c (#15) and are dynamically stable. They also display a clear metallic character. Substitution of Sc for Y (M2), however, leads to reduced bond strength for W–C and Al–Al interactions, while M2–C and M2–Al interactions are strengthened, resulting in slightly decreased moduli; from B = 143 GPa, G = 94 GPa, and E = 231 GPa to B = 130 GPa, G = 91 GPa, and E = 219 GPa, respectively. Extending the comparison to other Y-based i-MAX phases, the moduli increases along the series where M1 = Cr, Mo, and W.
Acknowledgments
The calculations were carried out using supercomputer resources provided by the Swedish National Infrastructure for Computing (SNIC) at the National Supercomputer Centre (NSC), the High Performance Computing Center North (HPC2N), and the PDC Center for High Performance Computing. We acknowledge support from the Knut and Alice Wallenberg (KAW) Foundation for a Fellowship Grant and Project funding (KAW 2015.0043), and from the Swedish Foundation for Strategic Research (SSF), program EM16-0004.