

Abstract
In the last decade, attempts at synthesizing artificial cells for studying cell biology or for biomedical and therapeutic applications have increased dramatically. Yet, a large part of research is still dependent on conventional reconstitution techniques. To increase the level of complexity of artificial cell models, recent advances in droplet microfluidics pose tremendous potential for construction of artificial cells by providing a membrane interface that can harbor biochemical processes. In this review, we describe the growing field of artificial cell engineering with the focus on depicting advances in microfluidic approaches for encapsulation of biomolecules.
Export citation and abstract BibTeX RIS
Introduction
Ever since the first cells were observed by Robert Hooke [1], scientists have been fascinated by the sheer complexity of these micron-scale living entities that have evolved into highly efficient energy-utilization machines. With the development of technology to physically probe into the functioning of cells [2–4], a lot of fundamental characteristics akin to all cells have been discovered and cell functions such as migration, division, endocytosis and exocytosis, and many others are understood as essential cellular processes. However, the assembly of how component molecules overcome energetic and entropic barriers to mediate such macroscale phenomena within the complex design of cells has remained elusive over the years. The central dogma of cell biology elucidates the role played by nucleic acids and proteins in carrying out these cell functions. While modern microscopic techniques in combination with standard biochemical assays have made it possible to observe dynamics of molecules or to reconstitute enzymatic reactions, the fact remains that inherent interactions of protein(s) of interest with other molecules (could be thousands of other molecules in a cell's cytoplasm) present in the confined cell volume are not directly accounted for.
To a certain degree, there exist disparities in protein behavior in cells as compared to in vitro systems that attempt to recapitulate them in isolation. This can be in part attributed to the degeneracy and diversity in the functions of biomolecules [5]. To delineate the complexity associated with proteins and nucleic acids inside cells, researchers have devised strategies to reconstitute different aspects of cell functions in a stepwise manner similar to the additive manufacturing of cars or airplanes. This approach comes under the purview of bottom-up synthetic biology and one aspect of such controlled reconstitution is the construction of artificial cells [6]. It is important to note that an ideal definition artificial cell would be the simplest living entity that is not naturally present. David Deamer [7] has previously referred to the problem of origin of life and how construction of an artificial cell would require the reconstitution of several key features of basic cellular processes. However, given that we do not fully understand the underlying mechanism of molecular arrangement and interaction within cells resulting in its complexity, an alternative definition of an artificial cell could be a membrane-bound compartment capable of sustaining isolated biochemical reactions directed towards mimicking specific cellular functions. Droplet microfluidics, being the state of the art technology in many biomedical and therapeutic applications [8, 9], could provide a suitable platform for manipulating biomolecules in microscale confinements and thus enabling the construction of artificial cells [10].
Artificial cells
The prospect of designing an artificial cell was first proposed by Thomas Chang in 1957 when he showed molecular transport into compartments made from polymer porous membranes as an analogy to the movement of ions and water across the cell membrane [11]. Subsequent work led to realization of the importance of similar compartments containing bioactive compounds as potential therapeutic agents. Chang and others went on to propose their use as artificial red blood cells, for functional isolation of islets of endocrine cells, stem cells, genetically engineered cells and as delivery vehicles for drugs, enzymes and micro/nano biosensors [12, 13]. Since the motivation behind the construction of these artificial cells was for therapeutic applications, not much emphasis was placed on the components constituting these systems. Although mimicking certain cell functions, these artificial cells were neither structurally similar nor were they made with components found in real cells. One type of classification of artificial cells can be made based on the extent of their structural similarity to real cells in addition to desired functional reconstitution. As mentioned elsewhere [14], 'typical' artificial cells should be similar in structure and function to real cells with its constituents primarily extracted from real cells while 'non-typical' ones are only expected to mimic specific cellular functions without any restrictions on its molecular constituents or structure. Common approaches to reconstituting controlled cellular functions in artificial cells include using a top-down approach where undesirable parts are systematically removed or modified from what real cells do or a bottom-up approach where natural or synthetic molecules are assembled into a cell-like unit [15]. Any attempt at reconstituting an artificial cell should consider the creation of compartmentalized volume surrounded by a boundary to prevent direct interaction of the confinement space with its environment. This is important to ensure a non-equilibrium setting to mediate directed flow of necessary molecules and energy which has been efficiently mastered by real cells. The fluid mosaic model of the plasma membrane of cells proposed by Singer and Nicholson emphasized the importance of a membrane as an essential component for arranging proteins and selectively allowing the exchange of molecules necessary for sustaining cellular processes [16, 17]. In order for the artificial cells to simulate cell function, they need membranes to separate an inner solution containing essential biomolecules from its surroundings and provide a hospitable environment for proteins to organize and function appropriately. Furthermore, the inner core of such artificial cells should be capable of maintaining a sufficiently crowded environment for sustaining biochemical reactions through a continued energy regeneration process. In the next two sections, we review work on reconstituting cell-like membranes in artificial cells with encapsulated nucleic acids and proteins and discuss the wide variety of methods used to simulate cell function.
Membrane-bound compartments
Lipids are the major constituents of all cell membranes and are hypothesized to have formed the first membranes in primitive cells owing to their ability to self-assemble in bilayer vesicles in aqueous environment [18]. Phospholipid vesicles or liposomes have been predominantly used as compartment models for the bottom-up construction of artificial cells. The major advantage of a self-assembled lipid bilayer is its proximity in shape and property to cell membranes which is important for simulating the flexible yet continuous barrier of cells separating the cytoplasm from the outer environment. Depending on the type of lipids, the permeability of the membranes can be controlled. Bangham et al were one of the first to demonstrate diffusion of monovalent ions across a phospholipid bilayer [19]. Further studies showed the ability of the lipid bilayer to be permeable to protons and potassium ions based on their thickness which was modulated by incorporating long and short chain phospholipids [20]. Phase separation of short and long chain lipids within the membrane and the presence of lipid rafts, common phenomena associated with cell membranes and important for cell function, have also been observed in liposomal membranes [21, 22]. Reconstitution of functional membrane proteins in these liposomal membranes further strengthened their utility in mimicking the plasma membrane of cells. Several techniques of assembling liposomes have been developed over the years, a majority of which is discussed in detail in later sections. Another membrane candidate are fatty acid vesicles. Like liposomes, fatty acids were shown to self-assemble at critical concentrations into bilayer vesicles [23] and self-propel droplets along a concentration gradient [24]. While being able to confine and sustain oligonucleotide synthesis in its lumen, fatty acid vesicles were shown to be more permeable and demonstrated inhibitory effects towards transcription when compared to phospholipid vesicles [25, 26].
In order to increase the stability of artificial cell membranes, polymersomes or vesicles made of different types of polymers have been generated [27, 28]. This was primarily aimed at encapsulating drugs and other therapeutic agents within polymersomes for greater stability and targeted delivery. Membrane proteins were reconstituted in polymersomes with retention of function in some cases [29, 30]. However, such membrane activity was reduced in polymer membranes as compared to liposomes. Proteinosomes are other artificial cell membrane substitutes. They are made by conjugating proteins of interest (usually bovine serum albumin; BSA) with multiple chains of a polymer PNIPAAm and assembled at the interface of aqueous droplets. Like polymersomes, their predominant use has been towards encapsulating drugs for targeted delivery. Membrane permeability and membrane fusion using proteinosomes were demonstrated comparing their similarity to liposomal membranes [31]. Hybrid membranes made of phospholipids and diblock copolymers or amphiphilic short chain peptides have been shown to increase the stability of liposomes while retaining the flexibility and permeability of lipid bilayer membranes [32–34]. However, problems of hydrophobic mismatch in the bilayer, disintegration of lipid polymer interaction and improper membrane protein reconstitution require further investigation into their ability to sustain cell-like membrane activity [33].
Cytoplasm of artificial cells
As mentioned above, compartmentalization is essential to the functioning of an artificial cell. Once a compartment surrounded by an appropriate membrane or boundary is established, the cytoplasm needs to be defined precisely to reconstitute desired cellular processes. Since proteins serve as the workhorse of any cells, initial attempts at reconstitution were carried out using purified proteins. Cytosolic or soluble proteins are easier to encapsulate when compared to membrane proteins which require proper folding into an existing membrane for retention of functionality. Given the molecular complexity and functional diversity of the cytoplasm of cells, reconstituting the cytoplasm of an artificial cell can be a three-part process: building the cytoskeleton, replication of genetic material, and reconstitution of enzyme-linked biochemical reactions.
Several studies have reported the reconstitution of actin filaments in liposomes as first models of artificial cell cytoskeleton [35]. Pontani et al demonstrated the formation of an action cortex like structure inside liposomes encapsulated with purified actin and other nucleation and anchoring proteins [36]. Kaufmann et al observed the nucleation of actin filaments at lipid membranes by embedded talin [37]. Further, changes in liposomal shapes with actin polymerization in the presence of crosslinking proteins such as fascin, filamin, alpha-actinin and heavy meromyosin were demonstrated [38]. These effects suggest the possibility of mimicking changes in cell shape and size in response to external stimuli such as adhesion to a surface or migration in the direction of a chemokine gradient. Likewise, microtubule encapsulation inside liposomes resulted in microtubule growth and buckling causing extension of the liposomal membrane in the direction of growth [39, 40]. Other studies have reported similar effects in liposomes using intermediate filament proteins such as vimentin [41].
For mimicking replication of genetic material and subsequent delivery into daughter cells, it is important to carry out oligonucleotide synthesis in the lumen of the artificial cell compartment. A straight forward approach is to encapsulate necessary components of a polymerase chain reaction with primers and a DNA template to validate amplification in the confined volume. A similar experiment was reported by Kurihara et al where DNA amplification inside liposome triggered fission in the presence of a bolamphiphilic compound (an amphiphilic molecule that has hydrophilic groups at both ends of a hydrocarbon chain) into daughter artificial cells with DNA transfer [42]. A subsequent publication further demonstrated the ability of these liposomes to carry on DNA synthesis and replication for two generations by adding excess reagents via vesicle fusion [43]. A self-sustained DNA synthesis inside lipid vesicles using rolling circle amplification while encoding DNA polymerase was shown in a separate study [44], suggesting the possibility of reconstituting autocatalytic reaction.
Finally, the third part forms the bulk of the cytoplasm which is essentially a mix of a wide variety of proteins, enzymes, ions and other biomolecules required for mediating protein synthesis and biochemical reactions involving protein-protein interactions. The major advantage of using purified proteins is that relative concentrations of encapsulated proteins can be well controlled. Since recombinant protein purification usually yields high concentrations of proteins, they can be easily diluted to achieve a range of working concentrations. However, membrane protein reconstitution remains challenging when using purified proteins [45]. Nevertheless, many studies of artificial cell construction were carried out using purified cytosolic and membrane proteins. An alternative approach to reconstitute necessary proteins in the cytoplasm of artificial cells which has become popular in recent years is by using cell-free expression (CFE) systems [46]. These are cell-extracts containing ribosomes, transcription factors and amino-acyl synthetases supplemented with charged nucleotides, amino acid mixture and an energy regeneration mix. The modularity of CFE system allows expression of multiple proteins simultaneously from recombinant plasmids. Past studies have shown expression and functional reconstitution of different kinds of membrane proteins such as ion channels [47, 48], mechanosensitive channels [49, 50], G protein-coupled receptors [51], enzymes for lipid synthesis [52, 53], olfactory receptors [54], gap junction proteins [55], proton pumps [56] and transporter proteins [57, 58]. Also, it is possible to incorporate post translational modifications in proteins using eukaryotic CFE [59]. One drawback of these systems is the variation in protein expression that depends on the confinement volume [60]. Also, competition for resources inhibits transcription and translation of proteins under similar promotors [61] making it difficult to control relative proportions of multiple proteins.
Artificial cell assembly
While different polymers, such as poly(lactide-co-glycolide), have been used to encapsulate biological materials, lipids best represent the cell membrane from both functional and structural perspective. In the assembly of artificial cell components, the lipid vesicle plays an integral role in mimicking the cell membrane and its functions in creating an insulated niche for the sub-component cellular machineries while simultaneously allowing fluxes of intracellular and extracellular molecules in response to internal and external signaling. Giant unilamellar vesicles (GUVs), made up of a single phospholipid bilayer with compositions that one desires, are currently the best representatives in chemically, structurally and dimensionally mimicking a natural cell membrane [17]. Below, we briefly describe some common approaches in making vesicles made of lipids to mimic cell membranes.
Hydration
The formation of GUVs by hydrations goes as far back as 1969 and is one of the first methods used for forming GUVs [62]. In this assay, phospholipids films are formed on a substrate by dehydrating solvents such as chloroform. Then, these films are rehydrated in aqueous buffer, and allowed to swell from a couple of hours to a couple of days [45, 62, 63]. Although the hydration method has been used in many studies, such as for membrane mechanical property characterization, membrane dynamics, protein interaction and vesicle fission [64], there are drawbacks in this approach. These include inevitable formation of multilamellar vesicles, low encapsulation efficiency, size inconsistency, and long hydration time.
Electroformation
An advanced and more efficient version of the hydration method, electroformation uses electric fields to form GUVs [65]. Similar to the hydration process, solvent (chloroform/methanol mix) dissolved phospholipids are spread on parallel electrodes and solvents are allowed to evaporate. A solution of interest (generally low ionic strength) is added and AC electric fields are applied which consequently facilitate rapid swelling of lipid bilayers and result in the formation of GUVs [66]. Although there is a higher yield (80%) of unilamellar vesicles compared to the hydration method (40% yield), the shortcomings of electroformation include potential oxidation of polyunsaturated lipids, low encapsulation efficiency and size inconsistency. Also, it is not possible to effectively encapsulate complex biological solutions such as CFE reactions using electroformation as the vesicles simply do not swell [67].
Vesicle fusion
Another method of making GUVs is the method of vesicle fusion [63]. The idea behind this method is fusing small unilamellar vesicles (SUVs) or large unilamellar vesicles to form GUVs. Dried lipid films on a substrate are rehydrated slowly to form a mixture of multilamellar vesicles (MLVs) which is then extruded through a porous polycarbonate membrane which extrudes the MLVs into a SUVs by physical shearing. However, this method is demanding as it requires utilizing components to supply the energy to open up SUVs and initiate fusion [64]. Most commonly used approach for vesicle fusion to form GUVs are peptides that bind together to form functional proteins such as SNARE proteins, and this method is commonly known as peptide-induced fusion [45, 68]. The SUVs can also be used to fuse onto a larger vesicle made by electroformation/hydration or used as artificial cell models for the reconstitution of membrane proteins. Others have also reported a method by which fusion is facilitated by phospholipids of opposite charge [63]. Moreover, some have reported making GUVs from SUVs as a spontaneous result of leaving SUVs inside a buffer solution for a long period or by cycling of freezing and thawing of SUV-containing solutions [64].
Although predominantly used for making lipid bilayer vesicles, the techniques above can be extended to the generation of polymersomes, proteinosomes and hybrid membranes given the amphiphilic nature of the constituent molecules in each case. Despite their popularity and relative ease of implementation compared to the more modern methods, the limitations mentioned above render them inappropriate for the construction of more complex artificial cells. Hybrid approaches where GUVs are first made by hydration or electroformation and then fused with SUVs containing membrane proteins of interest require the use of detergents, which can cause protein aggregation and also increase the ionic permeability of the membranes that can affect reconstitution of cellular processes requiring absence of certain ionic species [45]. Nonetheless, these conventional approaches have been commonly used in recent studies of reconstitution of biochemical reactions in artificial cell models [69–74].
It is evident that there is a wide scope for development of reliable and efficient methods for the synthesis of membrane-bound compartments with encapsulated biological solutions that would recapitulate desired cellular functions. With an increasing need for reconstitution of a multitude of interacting proteins in vitro, more modular and stepwise control in the bottom-up assembly of artificial cells is desirable. Over the years, concerted efforts have been made at resolving this technical challenge using novel microfluidic devices. As described in other review articles in recent years [8, 75],droplet microfluidics is a promising tool for the study of confined biochemical reactions with capabilities such as detection and sorting of individual droplets, picoinjection for mixing different reaction species, emulsion creaming for long term uniform incubation and modularity in design allowing it to be coupled with standard quantitative assays such as flow cytometry and mass spectrometry. Nanolitre and picolitre droplets can serve as an ideal size range for artificial cell volumes. Since microfluidic devices ensure high throughout generation of droplets, high yield is an added advantage. Further, the ability to sequentially add and mix reagents supports the basic philosophy of bottom-up assembly. Thus, for the construction of complex artificial cells, droplet microfluidic devices can serve as a technological leap [10, 76]. In the next section, we review droplet microfluidic devices developed for the synthesis of artificial cells.
Constructing artificial cells using droplet microfluidics
As stated earlier, lipids are natural constituents of cell membranes and exhibit a wide range of physical behaviors such as phase separation, raft formation and leaflet asymmetry that are desirable for proper organization and functioning of membrane proteins. Here we focus on mainly microfluidic techniques where lipids were predominantly used as the membranes for artificial cells. Certain studies with polymersomes and peptide-based membranes are discussed as well owing to the use of droplet microfluidics in those studies.
Droplet encapsulation
Although compartmentalization is essential for segregation of reaction species, a fully formed membrane is not necessary for sustaining simple biochemical reactions. The simplest artificial cells can be purified proteins or cell-free lysate and expression systems encapsulated within aqueous droplets in an immiscible oil with or without an interfacial leaflet of lipids. The lipids help stabilize the droplets due to their amphiphilic property and also allow headgroup interaction with encapsulated molecules which has been shown to play a role in protein expression using cell-free systems and for ensuring spatial localization of membrane and soluble proteins [60]. These droplets can be readily generated with uniform size distribution using a microfluidic device with the oil and water phases flowing towards a converging junction (T-junction) [77] (figure 1(A)). Alternatively, vortexing is a conventional approach towards generating polydisperse droplets. Guan et al reconstituted cell-cycle oscillations in aqueous droplets of a range of sizes containing cell-free xenopus egg extracts [78]. Demembranated sperm chromatin was also added to induce self-assembly of nuclei followed by chromatin decondensation timed by the mitotic oscillator. Whole cell encapsulation in a hydrogel has also been considered as an artificial cell model [79, 80]. As an example, Tan and Takeuchi used a T-junction droplet generating device to make monodisperse alginate hydrogel droplets encapsulating Jurkat cells [81]. Alginate hydrogels provide immunoisolation for transplanted islets of cells while allowing necessary molecules to diffuse into the droplets for cell sustenance. Another aspect of droplet-based artificial cells with spatial localization of reaction species by liquid–liquid phase separation (LLPS) was previously demonstrated [82]. Interestingly, Sokolova et al found enhanced transcription rates in coacervates induced in droplets encapsulating CFE solution with varying concentrations of polyethylene glycol (PEG) [83]. This suggests the possibility of membrane-less compartmentalization of biochemical reactions within droplets as primitive models for artificial cell synthesis. Although single emulsion droplet encapsulation is a well-established method and a simple approach for encapsulation into cell-sized compartments, the lack of lipidic membrane prevents engineering of membrane-based cellular processes.

Figure 1. (A) Images showing the encapsulation of a HeLa based CFE solution in aqueous droplets. 2% Span 80 was used in mineral oil for stability of droplets. Scale bar: 100 µm. (B) Droplet transfer method of making lipid bilayer vesicles using the conventional approach as shown on left and the microfluidic assembly line shown on right. Reprinted with permission from [133]. Copyright (2011) American Chemical Society, (B) left. Reprinted with permission from [85]. Copyright 2011 American Chemical Society, (B) right. (C) Double emulsion templated vesicle formation using glass capillary based microfluidic device. Images in bottom show the dewetting of oil droplet containing lipids from the double emulsion resulting in liposome formation. Reprinted with permission from [113]. Copyright 2016 American Chemical Society.
Download figure:
Standard image High-resolution imageInverse emulsion method
With the merits of encapsulating biological solutions within membrane-bound compartments in mind, the next innovation in artificial cell synthesis was the use of inverse emulsion transfer to generate lipid bilayer vesicles. This is the most common method of encapsulating purified proteins and CFE systems within vesicles used till date. Early studies by Pautot et al demonstrated the formation of vesicles using inverse emulsion [84]. Briefly, the solution to be encapsulated is dispersed in an oil phase containing lipids. Vortexing generates polydisperse droplets that are then laid on top of an outer aqueous solution to form a lipid monolayer interface by diffusion from the oil phase. Finally, density-based centrifugation allows the droplets to cross the interface forming lipid bilayer vesicles in the process. By using a T-junction device, Matosevic et al integrated the two-step process in a single step microfluidic device using a triangular object to direct droplet transfer by flow across the oil aqueous interface (figure 1(B)) [85]. Typical vesicle sizes (~20–70 µm) are determined by the size of the channels of the device and the flow rates used to form droplets. Similar device was also developed by Karamdad et al where they carried out droplet generation and transfer across oil-aqueous interface in a sequential manner and further estimated bending rigidity of the lipid vesicles using fluctuation analysis [86]. It is important to note here that this is one of the few methods of making bilayer vesicles exhibiting membrane asymmetry between the inner and outer leaflets. Pautot et al demonstrated this process by using different lipids and diblock copolymers for making the droplets and the oil aqueous interface [87]. A 99:1 solution of dodecane:silicone oil was used to dissolve the inner leaflet lipids while the outer leaflet lipids were dissolved in silicone oil to ensure density-based separation of the two oils and prevent lipid mixing. As shown in subsequent studies, membrane asymmetry does affect mechanical properties of lipid bilayer vesicles [88, 89]. The primary advantage of using this approach is the flexibility in terms of the inner solution that is encapsulated in the vesicles. Several studies have shown encapsulation of bacterial and eukaryotic CFE systems as well as purified protein systems such as the cytoskeletal proteins discussed earlier [36, 90–92]. Past studies have demonstrated the reconstitution of functional membrane proteins like bacteriorhodopsin and photosynthetic reaction centers [71], alpha-haemolysin [93] and mechanosensitive channels in liposomes [91] by using this approach.
There are growing interests in the phenomena of LLPS as an organizing principle in cells where two or more polymeric solutions in the same phase can separate into self-assembled droplets with a diffusion accessible interface when the concentrations are sufficiently high [82, 94]. One such system using the bacterial cell division protein FtsZ and PEG (polyethylene glycol) 8/dextran 500 in an aqueous phase was reported where Sobrinos-Sanguino et al encapsulated this solution inside Escherichia coli lipid-based bilayer vesicles and studied their LLPS behavior [95]. This was referred to as a cytoplasm-mimic with filamentous and soluble components in the interior of the vesicles. Abkarian et al demonstrated a modified technique of generating encapsulated liposomes by setting up the inverse emulsion assembly in a centrifuge enabled platform [96]. Using this assembly called cDICE (continuous droplet interface crossing encapsulation), liposomes encapsulated with the bacterial protein MinD were shown to induce oscillations in fluorescence along their membranes mimicking the protein's natural oscillation in bacterial cells [97]. One potential drawback of this approach is that there is inherent leakiness of the membranes to small molecules such as IPTG (Isopropyl β-D-1-thiogalactopyranoside) (unpublished results). Finally, different oil phases are necessary for use of different kinds of lipids to ensure high yield of vesicles since the interface formation is highly dependent on the types of lipids used [98] (unpublished observations).
Double emulsion templated vesicles
Generation of double emulsion templated polymer vesicles were first shown by Lorenceau et al using a glass capillary-based microfluidic flow focusing device (Figure 1(C)) [99]. As the name suggests, double emulsions require at least three phases and two interfaces to form. Till date, several variants of the glass capillary device have been reported with similar flow features and formation of double emulsions. Glass devices are used due to the need to use organic solvents for dissolving lipid molecules which have negative effects on materials like PDMS (polydimethyl siloxane). Since most non-membrane proteins are soluble in aqueous phase, double emulsions with water/oil/water phases are made by coflowing the inner aqueous phase with an oil phase containing the membrane molecules towards a flow focusing junction surrounded by the outer aqueous phase. Shum et al demonstrated the formation of phospholipid vesicles using this approach by dissolving lipids in an oil phase of chloroform and toluene [100]. Later improvements resulted in an updated version of the device wherein ultrathin double emulsions were made with lipids dissolved in a mixture of chloroform and hexane to facilitate dewetting [101, 102]. A wide variety of lipid mixtures can be dissolved and used to form phospholipid vesicle membranes while displaying lipid bilayer properties such as phase separation and lipid microdomains [103].
Using diblock copolymers such as PEG-b-PLA (polyethylene glycol-b-poly-L-lactide), multi-compartment vesicles were also made by this method showing their potential use as artificial cell organelles for internal organization of proteins [104, 105]. Given the flexibility of using different types of inner solution for encapsulation, our lab and others adapted this device for encapsulating CFE reactions and purified proteins in artificial cells using lipids or polymer-based membranes [91, 101, 106, 107]. Martino et al showed successful encapsulation of an E. coli-based cell-free reaction solution with plasmid containing the gene for MreB-RFP (red fluorescent protein) [108]. MreB is a bacterial filamentous protein that is known to traverse the bacterial membrane in a helical manner to ensure rod-shaped growth of the bacterial cells [92]. They used PEG-b-PLA along with additional PLA homopolymers to form polymersomes using a mixture of chloroform and hexane as the middle phase. Upon incubation, red fluorescent puncta were observed initially with the MreB localizing to the polymer membrane over time owing to the hydrophobicity of PLA homopolymers. Finally, MreB-RFP release from the interior into the outer solution was observed through pores in the membrane which were formed when the vesicles were exposed to hypoosmotic shock.
Further use of this glass capillary device was illustrated in the formation of artificial cells which displayed LLPS in their cytoplasm-mimicking a membrane-less artificial nucleus [109]. Briefly, coacervates made from an E. coli CFE system and polyuridylic acid (polyU)/spermine were encapsulated along with added DNA encoding for Spinach2 aptamer which binds to 5-difluoro-4-hydroxybenzylidene imidazolinone (DFHBI) to form a fluorescent complex. Since DNA was shown to localize with the coacervates, CFE of Spinach2 was only carried out in the LLPS domain resulting in bright fluorescence in a small spot within the vesicle. Interestingly, these coacervates show reversible phase transition in response to changes in temperature. When the temperature was decreased below the lower critical solution temperature (LCST) the coacervates dissolved resulting in diffuse fluorescence in the lumen of the encapsulated liposomes.
Our lab has also demonstrated the construction of artificial cells using a similar version of the glass capillary device. Caschera and Lee et al demonstrated the use of this device to form artificial cells capable of expressing in vitro synthesized ribosomes from rRNA (ribosomal RNA) operon and their subsequent utilization in the synthesis of a super-folder green fluorescent protein (sfGFP) [110]. An E. coli crude extract devoid of ribosomes was encapsulated along with plasmids containing the rRNA operon and the gene for sfGFP. Upon incubation of the liposomes in a feeding solution to allow membrane permeability of small energy molecules, sfGFP fluorescence was detected within the lumen of the vesicles suggesting sequential CFE of ribosomes and sfGFP [109]. We showed CFE of MscL (mechanosensitive channel of large conductance) in a HeLa cell-based extract [107]. The MscL localized to the membrane of the vesicles which was visualized by expressing MscL-eGFP (enhanced GFP). 2% of polyvinyl alcohol (PVA) was necessary for stability and formation of double emulsions encapsulating HeLa-based CFE using this device. However, we found that PVA generated protein aggregates in the cell-free reactions at the concentration levels necessary for double emulsion generation [111]. In a subsequent study, we used an E. coli cell-free expression system to create artificial cells capable of expressing both cytosolic and membrane proteins simultaneously with different functionalities [91]. One protein was G-GECO which is a calcium biosensor that fluoresces when calcium ions are bound. The other expressed protein was MscL which is a membrane protein and formed a non-selective transport channel upon increase in membrane tension of the lipid vesicles. When exposed to a calcium-containing hypoosmotic solution, an increase in membrane tension due to swelling resulted in MscL activity and subsequent increase of G-GECO fluorescence. Thus, an artificial cell capable of interacting with its surroundings via coupled signaling was demonstrated.
By using a diblock copolymer (F-68) at varying concentrations, Deng et al showed reversible shrinking of liposomes with attached lipid droplet when hyperosmotic solutions containing different concentrations of PEG were introduced [112]. Further, they demonstrated enhanced CFE of eGFP in shrunk vesicles due to increased macromolecular crowding. When the liposomes were generated with encapsulated PEG along with the E. coli CFE system, shrinking of vesicles resulted in formation of LLPS droplets of PEG in the lumen of the liposomes. This work showed the possibility to direct protein synthesis and segregation by controlling size of artificial cells. Other studies of vesicle formation using this glass capillary device demonstrated the possibility of generating multi-compartment artificial cells with proper control over each encapsulants. The device has been modified with two inner capillary tubes for simultaneously encapsulating multiple droplets within the double emulsions [113]. Upon careful control of flow rates and controlled dewetting in the presence of F-68, multi-compartment liposomes were formed which could contain two different solutions mimicking artificial cell organelles. Similarly, by concatenating multiple devices, the formation of vesosomes (liposomes within liposomes) containing CFE reactions with different plasmids was demonstrated to show segregated protein expression within an artificial cell [114].
Given the successes of this glass device for making double emulsion templated vesicles, polymersomes and liposomes [106, 100], and further improvements have been made to translate this approach into a conventional PDMS-based device. Thiele et al developed the first such device for making polymersomes using PEG-PLA copolymers dissolved in a chloroform/hexane solution [115]. In order to make PDMS amenable to the use of chloroform, a glass-like coating using sol-gel chemistry was implemented which allowed the preferential treatment of the inner and middle phase channel to be hydrophobic and the outer channel to be hydrophilic. Given the difficulty of making these glass-coated PDMS devices, a layer by layer deposition approach was used in a subsequent study to render the outer channel hydrophilic for the generation of W/O/W double emulsions [116]. Poly(allylamine hydrochloride) (PAH) and poly(sodium 4-styrenesulfonate) (PSS) solutions were flushed sequentially through the outer channels of the PDMS device while preventing it from entering the other channels by flowing water though them. Results indicated successful formation of double emulsions with 2% EA surfactant (polyethylenoxide-perfluoropolyether block-copolymer) in FC-40 oil. Deshpande et al devised a novel technique for making liposomes using a similar PDMS device without the use of volatile solvents such as chloroform and hexane which are incompatible with PDMS [117]. They used octanol as the middle phase for resuspending lipids and demonstrated rapid dewetting of the octanol lipid droplets, thus leaving a high yield of liposomes. Reconstitution of alpha-hemolysin that resulted in leakage of a fluorescent dye provided evidence supporting the formation of lipid bilayer membranes and very low octanol retention, if any. 5% PVA (30 000–70 000 M.W.) was used to coat the surface of the outer channel for hydrophilicity treatment and air was used to prevent the liquid from entering the inner and middle phase channels. The sizes of the liposomes were shown to be very uniform and can range from 5 to 20 µm. Another potential development of double emulsion device is the adoption of 3D-printed nozzle using 2-photon polymerization [118]. The nozzles have submicron resolutions and may be incorporated into PDMS devices to provide 3D nozzles that better resemble pulled glass capillaries.
One of the characteristics of double emulsion templated vesicles is oil retention in the middle phase and subsequent dewetting into a lipid reservoir droplet due to solvent evaporation [101, 102]. The existence or absence of oil after dewetting remains to be carefully characterized. Although the traditional double emulsion generating devices were incapable of attaining lipid membrane asymmetry, Lu et al devised a microfluidic technique to expose single emulsion droplets sequentially to lipid phases containing inner and outer leaflet lipids to form double emulsions which resulted in asymmetric liposomes [119]. Finally, while most studies have successfully implemented these devices for the synthesis of double emulsion templated liposomes and polymersomes, the compatibility of double emulsion generation with a range of biological solutions is still challenging and requires further investigation [120, 121].
Pulsed jetting
Another method of constructing artificial cells is by pulsed jetting of bilayer membranes. Funakoshi et al were the first to demonstrate this technique by blowing compressed air intermittently through a glass capillary containing an aqueous solution towards a planar lipid bilayer membrane [122]. A lipid bilayer can easily self-assemble at droplet interfaces. Upon jetting, akin to blowing soap bubbles, lipid vesicles were formed due to the shearing force of the liquid coming out of the glass capillary [123]. The biggest advantage of this approach is the ability to encapsulate any solution of interest irrespective of their complexity and physical properties. Also, it is possible to generate vesicles with bilayer leaflet asymmetry owing to the formation of a droplet interface where each droplet can contain different lipids. However, they reported significant oil between the lipid bilayers of these vesicles resulting in lower stability. Stachowiak et al improved the setup by using an inkjet nozzle in place of a microdispenser for pulsed jetting [124]. They were able to generate giant lipid vesicles around 200 µm in size. Also, encapsulation of monomeric actin and formation of filaments inside the vesicles were shown. SUVs reconstituted with v-SNARE proteins were encapsulated into the vesicles containing purified t-SNARE proteins. SNARE proteins are known to mediate lipid membrane fusion by forming a SNARE complex of t-SNARE and v-SNARE proteins [125]. The encapsulated SUVs localized to the vesicle membrane and calcium-dependent membrane fusion was demonstrated as is observed in synaptic junctions of neurons mimicking exocytosis. Kamiya et al followed up with this technique to generate vesosomes by sequential pulsed jetting through two planar lipid bilayer membranes [126]. It was shown that generating giant lipid vesicles using this method resulted in retention of oil in the bilayer. In a subsequent paper, it was demonstrated that the formation of asymmetric and oil-free liposomes could be achieved by using pulses that resulted in the formation of long lipid membrane tubes (figure 2(A)) [127]. These broke up into large oil containing vesicles and satellite oil-free liposomes which could then be isolated and observed (figure 2(B)). Lipid flip-flop activity between the inner and outer leaflets was observed and reconstitution of a gap junction protein connexin43 and pore forming membrane protein on the liposomal membrane resulted in leakage of a fluorescent dye, indicating formation of a lipid bilayer. A recent modification of the technique was implemented in a rotating cell to generate liposomes with different pairs of inner and outer leaflet lipid compositions in a high throughput manner [128]. A most recent study using pulsed jetting showed the formation of proteinosomes with fungal hydrophobin HFBI which are amphiphilic proteins that can stabilize planar bilayers at droplet interfaces [129]. Although this is a powerful approach for the construction of asymmetric artificial cells with encapsulated proteins, the more sophisticated setup and low yield prevented widespread adoption of this technique.
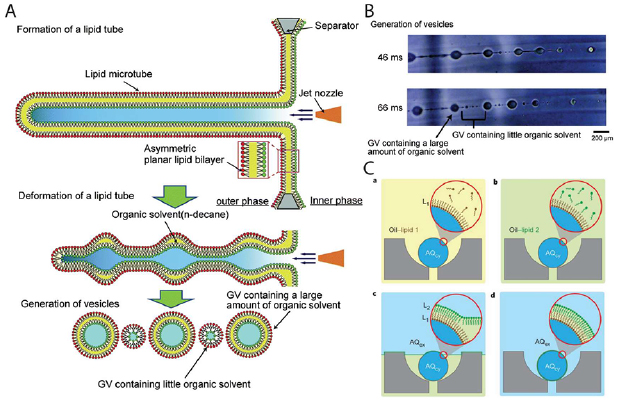
Figure 2. (A) Schematic depicting the formation of lipid tubes followed by onset of Rayleigh–Taylor instability resulting in the generation of large oil containing bilayer vesicles and small satellite bilayer vesicles with very little organic solvent. (B) Images of vesicle generation from jetting of lipid tubes. Scale bar: 200 µm. (A) and (B) [127] Copyright 2016 Springer Nature. With permission of Springer. (C) Layer-by-layer assembly of asymmetric lipid bilayer vesicles. (a) and (b) show sequential deposition of inner and outer leaflet lipids in oil phase. (c) and (d) show self-assembly and formation of vesicle when washed with aqueous phase. [130] Copyright 2013 Springer Nature. With permission of Springer.
Download figure:
Standard image High-resolution imageLayer-by-layer assembly
Another development towards generating asymmetric bilayer vesicles in recent years was made by Matosevic et al who introduced a microfluidic trapping device for layer by layer assembly of a lipid bilayer membrane (figure 2(C)) [130]. Aqueous droplets generated in an oil phase were trapped in cup-like structures followed by sequential washes with oils containing different types of lipids. Each passage of the aqueous oil interface across the droplet resulted in the formation of a monolayer of lipids. Subsequently, unilamellar and multilamellar vesicles could be made using this approach. The greatest advantage over other standard methods is the ability to generate asymmetric bilayer vesicles. However, given the low stability of the droplets during solvent exchange process and low probability of sustained entrapment of droplets, this clever approach of making asymmetric vesicles with encapsulated biological solutions has also not been widely adopted.
Microfluidic vesicle fusion at droplet interface
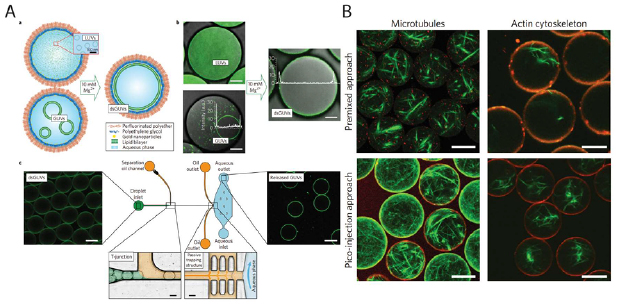
All the above-mentioned microfluidic techniques for bottom-up assembly of artificial cells require premixing of the cytoplasmic contents of the compartment before the final encapsulation step with lipid, polymer, protein or hybrid membranes. Recently, Weiss et al proposed a microfluidic platform for the assembly of lipid bilayer vesicles with modularity towards sequential encapsulation after the formation of the membrane (figure 3(A)) [131]. The droplet-based vesicles were named dsGUVs (droplet-stabilized GUVs) due to the use of copolymers in an oil phase for the generation of stable aqueous droplets. SUVs or GUVs made by extrusion or electroformation were encapsulated inside these droplets. At a high concentration of magnesium ions, the encapsulated SUVs and GUVs fused at the copolymer stabilized droplet interface to form a lipid bilayer membrane. Thereafter, microfluidic picoinjection was carried out to introduce a variety of biological solutions such as purified integrin IIbIIIa, RGD peptides, monomeric actin and microtubules inside the dsGUVs. During picoinjection, an electric field is applied to fuse droplets in contact which are otherwise highly stable due to the presence of an interface stabilizing molecule (e.g. perfluorinated polyether—polyethylene glycol). Thereafter, the dsGUVs were washed with a separation oil and flowed into an aqueous buffer. Functional reconstitution of integrins by binding of the released liposomes to a fibrinogen-coated surface was demonstrated. Actin cytoskeleton and microtubules were also reconstituted in the lumen of these liposomes (figure 3(B)). It was reported that the lipid bilayer formation in the presence of actin and tubulin was hindered when the copolymer stabilized droplets were made from premixed solutions of SUVs, GUVs and the purified proteins. Given the modularity of the microfluidic platform and the high yield of the generated liposomes, this device could have tremendous potential in the sequential encapsulation of complex biomolecules for step by step reconstitution of a cellular process. Although this approach seems quite versatile, the potential drawbacks include the complexity of the device and the sensitivity of lipid bilayer formation to magnesium ions.
{kind=link}
{kind=link}
Figure 3. (A(a)) Schematic showing the formation of droplet stabilized giant unilamellar vesicles (dsGUVs) from fusion of encapsulated GUVs and LUVs in the presence of magnesium ions. (b) Fluorescence images of the dsGUVs before and after formation of the lipid bilayer at the droplet interface. (c) Image on left shows dsGUVs in the microfluidic device (schematic depicted in the center) prior to droplet transfer across an intermediate oil phase into an aqueous solution (image on right). Scale bars: 20 µm. (B) Reconstitution of actin and microtubule filaments in GUVs. A comparison between premixed encapsulation and pico-injection based sequential encapsulation. [131] Copyright 2018 Springer Nature. With permission of Springer.
Download figure:
Standard image High-resolution image{kind=link}
Droplet-shooting and size-filtration
When encapsulating CFE solutions, most microfluidic techniques require sample volumes in the range of 50–100 µl which is large considering the volume of cell-free lysates obtained from a single batch preparation. Morita et al demonstrated an ingenious method of generating high yield of encapsulated vesicles from very small sample volumes (0.5–2 µl) using a modified inverse emulsion transfer [132]. Glass capillary containing the biological solution is mounted on a lab-made stand which can fit perfectly inside a 1.5 ml Eppendorf tube leaving some space at the bottom. As in inverse emulsion transfer method, an oil phase containing lipids is laid on top of an aqueous phase. The glass capillary mount is placed inside the tube and the assembled tube is then centrifuged. Small droplets of the loaded sample shoot out of the glass capillary and pass through the oil aqueous interface to form lipid bilayer vesicles. Use of two oil phases with different lipids and sequential droplet shooting can generate asymmetric liposomes which is a major advantage of this method. Drawbacks of this technique include size heterogeneity and possible change in molecular concentrations due to evaporation during droplet transfer through air.
Conclusion
Compartmentalization of biochemical reactions is critical to artificial cell research. We have described how various microfluidic approaches have aided the construction of artificial cells. Despite the rapid and promising progress to date, robust encapsulation of complex solution in cell-sized lipid bilayer vesicles remain a formidable challenge to many groups. There is a need for concerted efforts towards continued development of the technology to synthesize and manipulate cell-sized droplets or vesicles in the hope of developing better ways to assemble the molecules of life. It will require detailed and interdisciplinary understanding that include lipid biochemistry, interfacial chemistry, fluid mechanics, and microfluidic designs.
Acknowledgments
SM is supported by NSF-MCB 1561794. NW is supported by NSF-CBE 1844132. APL is supported by NSF-MCB 1561794, NSF-MCB 1817909, and NSF-CBE 1844132.