

Abstract
A comparative study of four series of pyrrhotite-type chalcogenide compounds Fe7−yMyX8 (X = S, Se) with substitution of Ti or Co for iron has been performed by means of x-ray and neutron powder diffraction, and by magnetization measurements. In Fe7−yMyX8 compounds having a ferrimagnetic order at  , the substitution of either Ti or Co for iron is observed to result in a monotonous decrease of the magnetic ordering temperature, while the resultant magnetization shows a non-monotonous behavior with a minimum around
, the substitution of either Ti or Co for iron is observed to result in a monotonous decrease of the magnetic ordering temperature, while the resultant magnetization shows a non-monotonous behavior with a minimum around  –1.5 in all the Fe7−yMyX8 families except Fe7−yCoySe8. Suppression of a magnetically ordered state with substitutions in Fe7−yMyX8 is ascribed to nearly zero values of Ti and Co magnetic moments, while the non-monotonous changes of the resultant magnetization are explained by the compensation of the sublattice magnetizations due to the non-random substitutions in alternating metallic layers. The difference in the cation partitioning observed in Fe7−yTiyX8 and Fe7−yCoyX8 is attributed to the difference in the spatial extension of Ti and Co 3d orbitals. High coercive field values (20–24 kOe) observed at low temperatures in the Ti-containing compounds Fe7−yTiyX8 with y ⩾ 3 are suggested to result from the enhancement of Fe orbital moment due to the Ti for Fe substitution.
–1.5 in all the Fe7−yMyX8 families except Fe7−yCoySe8. Suppression of a magnetically ordered state with substitutions in Fe7−yMyX8 is ascribed to nearly zero values of Ti and Co magnetic moments, while the non-monotonous changes of the resultant magnetization are explained by the compensation of the sublattice magnetizations due to the non-random substitutions in alternating metallic layers. The difference in the cation partitioning observed in Fe7−yTiyX8 and Fe7−yCoyX8 is attributed to the difference in the spatial extension of Ti and Co 3d orbitals. High coercive field values (20–24 kOe) observed at low temperatures in the Ti-containing compounds Fe7−yTiyX8 with y ⩾ 3 are suggested to result from the enhancement of Fe orbital moment due to the Ti for Fe substitution.
Export citation and abstract BibTeX RIS
1. Introduction
Iron-chalcogenide compounds with a composition around the equiatomic one have attracted great attention in recent years because of high-temperature superconductivity observed in FeSe-based compounds having a small Fe excess and a tetragonal crystal structure of the PbO-type [1]. A characteristic feature of these materials is the closeness of superconductivity to magnetism [2, 3]. Superconducting properties of bulk FeSe-based samples and films are strongly affected by substitutions, non-homogeneities, strains, by introducing vacancies and by the application of pressure [2–5]. A strong coupling between the crystal structure and magnetism was also revealed in Fe1−yX compounds (X = S, Se) having a deficiency of Fe and crystallizing in NiAs-type hexagonal structures [6]. The Fe57 Mossbauer [7] and x-ray emission spectroscopy studies of the hexagonal FeS [8] have revealed that applied pressure leads to the disappearance of a magnetic moment on iron atoms owing to the high-spin to low-spin transition at ~6.5 GPa. The collapse of the Fe magnetic moment under an applied pressure of about 5 GPa has been also observed in Fe7S8 (pyrrhotite) having a layered superstructure of the NiAs-type [7]. The layers containing chalcogen atoms in Fe7X8 (X = S, Se) are full, while among the layers containing iron atoms, one layer of the two is full whereas the other contains three atoms instead of four. The formation of superstructures in Fe7X8 (X = S, Se) is related to the different orderings of vacancies in alternating Fe layers. In particular, Fe7Se8 can crystallize in a triclinic 4C superstructure with the quadruplicated unit cell length along the c axis or in a trigonal 3C superstructure with the tripled unit cell dimension along the c axis in respect to the fundamental NiAs type cell. The 4C superstructure is a stable phase at room temperature, and can be obtained by slow cooling after low-temperature annealing, while the 3C superstructure is stable at high temperatures, but can be brought to room temperature by quenching [6, 9–14].
The compounds Fe7Se8 and Fe7S8 exhibit a ferrimagnetic order below the Neel temperatures of about (450–480) K [11, 12, 15–17] and 590 K [18–20], respectively. The magnetic moments of Fe in these compounds are arranged parallel to each other inside each layer, but are coupled antiparallel to each other between successive layers. Therefore, the presence of vacancies in every second metallic layer leads to incomplete compensation of magnetic moments and hence to the ferrimagnetism [12, 18]. The Co-based compounds Co7S8 and Co7Se8 having a reduced (~10%) interlayer distances in comparison with Fe7X8 are reported to be Pauli paramagnets [21, 22]. The substitution of Co for Fe in (Fe1−yCoy)7S8 and (Fe1−yCoy)7Se8 is found to result in the disappearance of a long range magnetic order at y ≈ 0.6–0.65 and magnetic moments on 3d metal ions [23–25]. There are two main reasons which are considered in the literature in order to explain the collapse of a magnetic moment on Fe atoms in Fe7X8-type compounds under applied pressure or owing to the Fe for Co substitution [7]: (i) the 3d band broadening due to the reduction of interlayer distances and overlapping 3d orbitals and (ii) the high-spin to low-spin transition due to changes in crystal electric fields.
The aim of the present work is to study the replacement effect of Fe atoms in Fe7S8 and Fe7Se8 by other 3d metal atoms on the crystal structure and magnetic properties. Our previous studies of Fe7−yTiyS8 compounds have revealed unusual non-monotonous change of the net magnetization with the Ti for Fe substitution, which was ascribed to the non-random distribution of Fe and Ti in cation layers [26]. In order to reveal the difference in the substitution of Fe by different 3d metal atoms in sulfur and selenium-based pyrrhotite-type compounds we have chosen titanium and cobalt as elements belonging to the first and second half of 3d series. Bearing in mind that using the traditional x-ray diffraction it is difficult to answer the question about partitioning of different 3d-metal atoms in the crystal lattice we have performed powder neutron diffraction measurements for some substituted compounds.
2. Experimental
Four series of polycrystalline samples Fe7−yTiyS8 (y = 0–4), Fe7−yTiySe8 (y = 0–3), Fe7−yCoyS8 (y = 0–7) and Fe7−yCoySe8 (y = 0–7) were synthesized by solid state reactions in evacuated quartz tubes. The starting materials were small pieces of 99.95% pure titanium, sulfur (99.99%), granules of 99.999% pure selenium, small pieces of cobalt (99.9%) and powder of iron (purity 99.98%). The tubes with starting materials were gradually heated in a furnace from room temperature up to 950 °C (with one-day temperature steps at 200, 400 and 600 °C), then the samples were kept for 2 weeks at temperature 950 °C. The obtained samples were ground, compressed in tablets, sealed in quartz tubes under vacuum and then homogenized in a furnace for another week at 900 °C. This homogenization treatment was repeated until obtaining single-phase samples. In order to examine the quality of the samples and the changes in the crystal structure upon substitution a powder x-ray diffractometer (Bruker D8 ADVANCE) with Cu Kα radiation was used. The measurements of the magnetic susceptibility and magnetization were performed by means of a Quantum Design SQUID MPMS magnetometer in the temperature interval 2 K ⩽ T ⩽ 350 K and Vibrating sample magnetometer Lake Shore VSM 7407 in the temperature interval 300 K ⩽ T ⩽ 1000 K. The samples for high-temperature measurements (above 300 K) were placed into evacuated quartz ampoules to prevent oxidation. The neutron powder diffraction (NPD) measurements were performed at room temperature using HRPT and DMC instruments at the Swiss Spallation Source SINQ with neutron wavelengths λ = 1.155 Å and λ = 2.458 Å, correspondingly. Refinement of the crystal structure has been done by means of the Fullprof program [27].
3. Results
3.1. X-ray data
Unlike the Fe7−yTiyS8 system in which the single phase samples were obtained up to Ti content y = 4 [26], we did not succeed in obtaining the Ti-substituted selenium-based Fe7−yTiySe8 samples with Ti concentrations above y = 3 even after prolonged heat treatments. As to the Co for Fe substituted systems, the all sulfide Fe7−yCoyS8 and selenide Fe7−yCoySe8 samples were determined by using x-ray diffraction to be single phase in the whole concentration range y = 0 – 7. As is turned out, the Ti for Fe substitution in the selenide compounds Fe7−yTiySe8 leads to the changes in the crystal structure analogous to that observed for the sulfur-based Fe7−yTiyS8 system [26]: i.e. from the initial 4С superstructure at y = 0, then to the 3C superstructure at y = 1 and to the monoclinic 2C superstructure with further increasing Ti content.
Figure 1 shows, as an example, the x-ray diffraction pattern for the Fe4Ti3Se8 compound from the selenide system. This diffraction pattern can be well described on the basis of a monoclinic 2C superstructure [a0 × a0 × 2c0] in the space group I12/m1 as for the sulfide compounds Fe7−yTiyS8 with high Ti concentrations (y > 1) (see [26, 28]) (a0, and c0—are the parameters of the primitive NiAs unit cell). The all Fe7−yCoySe8 compounds are observed to exhibit a trigonal 3C superstructure (P3121 space group). The Fe7−yCoyS8 samples with the Co content above y = 2 have the same superstructure, while the stacking faults are suggested to exist in the sulfide compounds with lower Co concentrations. It is worth to mention that although the substitutions of Ti or Co for iron in Fe7−yMyX8 leads to the changes in crystal structure and to the formation of different superstructures, all synthesized Fe7−yTiyX8 and Fe7−yCoyX8 compounds have a layered crystal structure of the NiAs-type and can be characterized by the lattice parameters a0 and c0 of the NiAs-type fundamental unit cell. In figures 2 and 3 we collected the data on variations of the lattice parameters a0 and c0 with substitutions in Fe7−yTiyX8 and Fe7−yCoyX8 systems. The results for the sulfide Fe7−yTiyS8 compounds are taken from our previous work [26].
× a0 × 2c0] in the space group I12/m1 as for the sulfide compounds Fe7−yTiyS8 with high Ti concentrations (y > 1) (see [26, 28]) (a0, and c0—are the parameters of the primitive NiAs unit cell). The all Fe7−yCoySe8 compounds are observed to exhibit a trigonal 3C superstructure (P3121 space group). The Fe7−yCoyS8 samples with the Co content above y = 2 have the same superstructure, while the stacking faults are suggested to exist in the sulfide compounds with lower Co concentrations. It is worth to mention that although the substitutions of Ti or Co for iron in Fe7−yMyX8 leads to the changes in crystal structure and to the formation of different superstructures, all synthesized Fe7−yTiyX8 and Fe7−yCoyX8 compounds have a layered crystal structure of the NiAs-type and can be characterized by the lattice parameters a0 and c0 of the NiAs-type fundamental unit cell. In figures 2 and 3 we collected the data on variations of the lattice parameters a0 and c0 with substitutions in Fe7−yTiyX8 and Fe7−yCoyX8 systems. The results for the sulfide Fe7−yTiyS8 compounds are taken from our previous work [26].
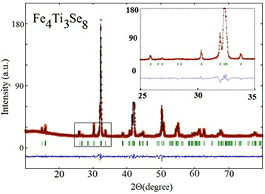
Figure 1. Observed (symbols) and calculated (line) x-ray diffraction pattern for Fe4Ti3Se8. Vertical bars indicate the Bragg peaks positions corresponding to the space group I12/m1 and the lattice parameters: a = 6.307(5) Å, b = 3.596(8) Å, c =11.845(7) Å, β = 90.6(7)°. The difference between calculated and observed intensities is shown in the bottom. Insert shows a part of the diffraction pattern in detail.
Download figure:
Standard image High-resolution image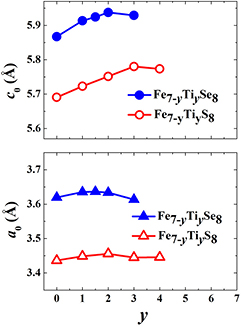
Figure 2. The lattice parameters a0 and c0 as a function of the Ti concentration in Fe7−yTiyS8 and Fe7−yTiySe8 compounds. The errors of the lattice parameters do not exceed the symbol sizes.
Download figure:
Standard image High-resolution image
Figure 3. The lattice parameters a0 and c0 as a function of the Co concentration in Fe7−yCoyS8 and Fe7−yCoySe8 compounds. The errors of the lattice parameters are within the symbol sizes.
Download figure:
Standard image High-resolution imageAs follows from figure 2, the Ti for Fe substitution up to y = 3 in the Fe7−yTiyS8 system and up to y = 2 in Fe7−yTiySe8 increases the c0 value which characterizes the average interlayer distances, while further growth of the Ti concentration does not affect substantially the crystal lattice. The substitution effect on the a0 parameter in Fe7−xTixX8 is less pronounced; the average intra-layer distance remains almost unchanged with increasing y. In the case of Fe7−yCoyX8 systems (figure 3), the growth of the Co content leads to the contraction of the crystal lattice in the direction perpendicular to the layers by about of 10.3% for Fe7−yCoySe8 and~8.9% for Fe7−yCoyS8, which is in agreement with published data [23, 24]. Unlike the c0 parameter, the a0 parameter is observed to be nearly constant in both the Co substituted systems. These results imply a substantially different compressibility of the crystal lattice perpendicular and parallel to the layers.
3.2. Magnetization of Fe7−yTiyX8 (X = S, Se) compounds
As follows from figure 4, which displays the temperature dependences of the magnetization of Fe7−yTiySe8 compounds, the initial Fe7Se8 compound exhibits the magnetic ordering temperature TN = 450 K, which consists very well with previously reported data [16]. The anomaly observed on the M(T) curve at temperature about 110 K is associated with the spin-reorientation transition [11, 12, 16, 29]. The Neel temperature of Fe7−yTiySe8 decreases with increasing Ti concentration from 450 K at y = 0 down to ~220 K at y = 3 (see inset in figure 4), which is indicative of the reduction of exchange interactions between Fe ions and suggests a low value or absence of a magnetic moment on Ti ions. Such a monotonous decrease of TN was also observed for Fe7−yTiyS8 [26] (shown by circles in the inset in figure 4). The sharp anomaly associated with the spin-reorientation transition is observed to disappear upon the Ti for Fe substitution in Fe7−yTiySe8. However, the samples with y = 1 and y = 1.5 show the reduction of the magnetization with decreasing temperature below 150 K and 300 K, respectively, which may be associated with gradual changes in the direction of Fe magnetic moments, i.e. with the 'smeared' spin-reorientation transition.

Figure 4. Magnetization of Fe7−yTiySe8 compounds with various Ti concentrations as a function of temperature in a magnetic field of 1 kOe. Insert shows the variation of TN with increasing Ti concentration for Fe7−yTiySe8 (triangles) and for Fe7−yTiyS8 (circles).
Download figure:
Standard image High-resolution imageAs follows from figure 4, unlike a monotonous reduction of TN the low-temperature (T ~ 2 K) magnetization of Fe7−yTiySe8 changes non-monotonously with increasing titanium content. Such non-monotonous change in the magnetization behavior with the substitution in Fe7−yTiySe8 is also evidenced by the field dependences of the magnetization measured at T = 2 K (shown in figure 5). Together with the data for the selenide system we plotted in figure 5 the field dependences of the magnetization for Fe7−yTiyS8 compounds (shown by doted lines). Some M(H) dependences for Fe7−yTiyS8 were taken from our previous papers [26, 28]. In both the selenide and sulfide systems Fe7−yTiyX8, the growth of the titanium concentration initially (up to y ~ 1.0 – 1.5) leads to a significant reduction and then to an increase of the magnetization with further substitution. However, there is a clear difference between the M versus H behaviors for sulfide and selenide compounds (see figure 5). An absence of the bulk spontaneous magnetization and a nearly linear M(H) dependence for Fe6Ti1S8 was considered in [26] as an indication for the full compensation of sublattice magnetizations, i.e. for an antiferromagnetic order in this compound. As follows from figure 5, unlike an antiferromagnetic behavior of the sulfide compound Fe6Ti1S8 the selenide compound with the same titanium content (Fe6Ti1Se8) exhibits a hysteresis loop and remnant magnetization. The presence of remanence in Fe7−yTiySe8 compounds with y ~ 1.0 – 1.5 shows that the sublattice magnetizations are not fully compensated probably because of a non-homogeneous distribution of Ti ions. All Ti-containing samples from the selenide system Fe7−yTiySe8 exhibit an enhanced coercivity in comparison with the sulfide family; however, the sulfide compounds Fe7−yTiyS8 with high Ti concentrations (y > 1.5) demonstrate substantially increased magnetization values in high magnetic fields. Variations of the magnetization in Bohr magnetons per formula unit, MFU, and coercive field at the Ti for Fe substitution in both systems are presented in figure 6. The values of MFU for Fe7S8 and Fe7Se8 (y = 0) measured in a field of 50 kOe at T = 2 K are found to be 2.08 μB and 2.28 μB, respectively, which is well consistent with reported data [30]; while for Fe7−yTiyS8, the magnetization reaches at y = 3 a value almost in twice higher than that observed for the non-substituted compound Fe7S8. An enhanced value of MFU for Fe4Ti3S8 is indicative of an appreciably decreased magnetization of one of the two sublattices, which together with the compensation at y = 1 implies the preferential Ti for Fe substitution in alternating cation layers [26, 28]. At high titanium concentrations, such a preferential substitution may lead to a reduction of the magnetic compensation in the ferrimagnetic unit cell of Fe7−yTiyS8 and results in an increase of the net magnetization. A less pronounced minimum of the magnetization at the Ti concentrations around y = 1 – 1.5 and a reduced MFU value for Fe7−yTiySe8 at y > 1.5 in comparison with the sulfide compounds (figure 6(a)) may be associated with the difference in the Ti distribution between layers in these systems as well as with the difference in the arrangement of Fe magnetic moments. Figure 6(b) shows the variation of the coercive field measured at 2 K in both sulfide and selenide systems with increasing Ti concentration. The Ti for Fe substutution leads to a non-monotonous growth of Hc values up to 21 kOe and 24 kOe for Fe4Ti3Se8 and Fe3Ti4S8, respectively.

Figure 5. Field dependences of the magnetization for Fe7−y TiySe8 (solid lines) and Fe7−y TiyS8 compounds (doted lines) at T = 2 K.
Download figure:
Standard image High-resolution image
Figure 6. Magnetic moment per formula unit in a field of 50 kOe (upper panel) and coercive field (lower panel) for Fe7−y TiyS8 (open symbols) and Fe7−y TiySe8 (full symbols) at T = 2 K as a function of the Ti concentration.
Download figure:
Standard image High-resolution imageIt is worth to mention that such high values of the coercivity are rarely observed in the compounds the magnetism of which is associated with Fe ions only. The enhancement of magnetic hardness in Fe7−yTiyX8 compounds at y > 2 may result from the growth of orbital moment of Fe atoms with the substitution. As to the non-monotonous behavior of Hc with the maximum in the vicinity of the Ti concentration y = 1.5, it presumably originates in a reduced resultant magnetization of the compounds with nearly compensated sublattice magnetizations [31]. As was observed in many ordinary ferrimagnets (see [32], for instance), the large values of the coercivity are usually observed around the compensation temperature.
3.3. Magnetization data for Fe7−yCoyX8 (X = S, Se)
Figure 7 shows temperature dependences of the magnetization measured at H = 1 kOe on the polycrystalline samples of Fe7−yCoyS8 and Fe7−yCoySe8 compounds. For both systems, the magnetic ordering temperature is observed to decrease monotonously with increasing cobalt concentration as in the case of the Ti for Fe substitution (see above). As follows from the inserts displayed in figure 7, the values of TN tend to zero at the critical cobalt concentration y ≈ 4.5, which is in good agreement with previously published data [23, 24].
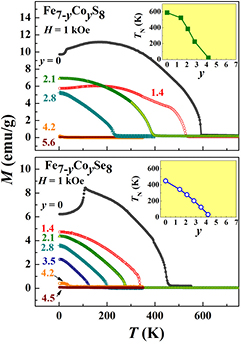
Figure 7. Temperature dependences of the magnetization for Fe7−y CoyS8 (upper panel) and Fe7−y CoySe8 (lower panel) systems measured at cooling in a magnetic field of 1 kOe. Inset show concentration dependences of the magnetic ordering temperature (TN) for both systems.
Download figure:
Standard image High-resolution imageSuch a behavior implies that the Co atoms replacing the iron atoms in Fe7−yCoyX8 compounds exhibit a substantially reduced magnetic moment and act as diluents relative to the magnetically ordered Fe subsystem. Indeed, from the NMR measurements performed on the Fe7−yCoyS8 samples [25], a value of the magnetic moment on Co atoms (µCo) can be estimated as ~0.4 μB at y ⩽ 3. At the Co concentration above y = 4, a magnetic moment on Fe and Co atoms was not detected in Fe7−yCoyS8 [25]. The influence of the Co for Fe substitution on the magnetization behavior under application of a magnetic field can be seen in figure 8 which displays the M versus H dependences measured on the Fe7−yCoyS8 and Fe7−yCoySe8 samples at 2 K. As follows from figure 8, the substitution of Co for Fe in both the Fe7S8 and Fe7Se8 compounds reduces the resultant magnetization; the spontaneous magnetization is not observed when the Co concentration exceeds y = 4. Unlike the Ti-substituted compounds, both the Co-substituted systems do not show large magnetic hysteresis in the magnetically ordered state even at low temperatures. The coercive field in these compounds decreases with increasing Co content up to y = 3; only around y = 3.5–4, some enhancement of the coercive field was observed (up to 4.5 kOe in the Fe7−yCoySe8 system at T = 2 K), which can be associated with the presence of magnetically non-homogeneous states near the critical concentration.
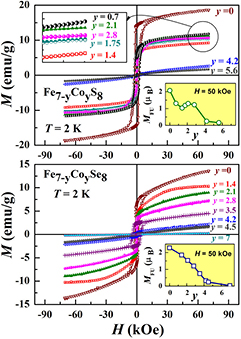
Figure 8. Field dependences of the magnetization measured at T = 2 K for Fe7−y CoyS8 (upper panel) and Fe7−y CoySe8 (lower panel) compounds with various Co concentrations. Insets show the concentration dependences of the magnetic moment per formula unit (MFU) measured at H = 50 kOe.
Download figure:
Standard image High-resolution imageHowever, as in the above considered Ti-substituted compounds, there is some difference in the magnetization behavior of sulfide and selenide samples with the Fe–Co substitution. A value of the magnetic moment per formula unit MFU for Fe7−yCoyS8 samples varies non-monotonously in the concentration range y = 0.7 – 2.8, while in the case of the Fe7−yCoySe8 system, the MFU monotonously decreases with increasing Co content (see insets in figure 8). The monotonous reduction of both TN and MFU in Fe7−yCoySe8 implies that the Co ions having a reduced magnetic moment substitute iron in both sublattices with near equal probability in whole concentration range (0 < y < 7), while in the case of Fe7−yCoyS8, some partitioning of Co ions apparently exists.
3.4. Neutron diffraction
In order to check our assumption about the non-random substitutions in Fe7−y My X8 compounds we have performed the neutron diffraction measurements at room temperature on powder samples of Fe4Ti3S8 and Fe4Co3S8 compounds having the same concentration of substituting atoms. According to magnetization data (see figures 4 and 7) these compounds are in the paramagnetic state at room temperature. As follows from analysis, the neutron powder diffraction (NPD) pattern for Fe4Ti3S8 can be well indexed by the 2C superstructure (space group I12/m1) as it was suggested for x-ray diffraction data in the work [28]. However, the Rietveld refinement was not able to get a nice agreement between observed and calculated NPD profiles within this model, which implies more complex ordering of Ti and Fe atoms over cation layers. Therefore, as a first step, the completely ordered monoclinic structure (space group C2/c) given by Powell et al [18] for the non-substituted Fe7S8 compound was used as a trial model for analysis of neutron diffraction data. It has been suggested that each of the four cation sites are equally occupied by Ti and Fe atoms (Fe3.5Ti3.5S8 stoichiometry).
The unit cell parameters, atomic coordinates, Ti and Fe occupation factors and profile function parameters were refined. The best fit results for the neutron diffraction patterns measured at λ = 1.15 Å and λ = 2.46 Å are shown in figures 9(a) and (b), correspondingly. Despite of the satisfactory agreement between observed and calculated profiles for the high-resolution neutron diffraction pattern measured with neutron wavelength λ = 1.15 Å (χ2 = 1.76, RB = 7.1%, Rwp = 4.17%, Rexp = 3.18%) one can see that high intensity data measured with neutron wavelength λ = 2.46 Å cannot be well fitted by the Powell's model. The superstructure peaks marked by (*) on the calculated profile are found to be absent on the experimental pattern (see the inset at figure 9(b)). This inconsistence drove us to search for another crystal structure model. The supergroup P2/c with in twice reduced unit cell parameters along the a and b directions in respect to Powell's model [18] has been chosen as a new trial model for Rietveld refinement. In comparison with completely ordered Powell's model which has normally vacant 4e Wyckoff site (0 y ¼) and fully occupied another 4e Wyckoff site (0 y' ¼), the higher symmetry model suggests that both of these sites are merged into one 2e Wyckoff site (0 y ¼) of the space group P2/c. In our trial model we suggested that the vacancies may be statistically distributed over one of two possible Wyckoff sites in deficient cation layers (2e or 2g Wyckoff site) by restriction the corresponding site occupation factor as much as 50%. Moreover, in the trial model each of the three cation sites were supposed to be equally occupied by Ti and Fe atoms representing Fe3.5Ti3.5S8 stoichiometry. Then the unit cell parameters, atomic coordinates, cation occupation factors and profile function parameters were allowed to be refined. The best fit result was obtained for half-filled Wyckoff 2g site for both neutron diffraction patterns measured at λ = 1.15 Å and λ = 2.46 Å (see figures 9(c) and (d), correspondingly). The schematic representation of the refined crystal structure is shown in figure 10.

Figure 9. The best fit result on the NPD patterns for Fe4Ti3S8 measured with neutron wavelength λ = 1.15 Å (a) and λ = 2.46 Å (b) by using the Powell's structure model [18], and λ = 1.15 Å (c) and λ = 2.46 (d) for P2/c structure model (see the text). Symbols are experimental values of the intensity and solid lines represent the results of the Rietveld refinement. The difference between calculated and observed profiles is shown at the bottom.
Download figure:
Standard image High-resolution image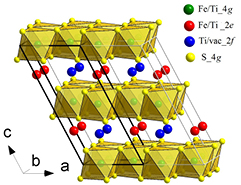
Figure 10. Schematic representation of the crystal structure refined from the neutron diffraction data for Fe4Ti3S8 (SG P2/c). The cations in fully occupied layers are located in the centers of MS6 octahedrons while the cations in layers with vacancies are shown as solid circles
Download figure:
Standard image High-resolution imageThe refined structural parameters as well as the agreement factors are given in table 1. As one can see from table 1, the fully occupied layers in Fe4Ti3S8 contain much more iron ions (Fe—76.6%) than titanium ions (Ti—23.3%) while the cation layers with vacancies are predominantly occupied with Ti atoms (Ti—53%; Fe—22.3%; vacancies—24.7%). Thus, the Rietveld refinement confirmed our suggestion based on magnetic measurements about the non-random Ti for Fe substitution in alternating cation layers.
Table 1. Structural parameters of Fe4Ti3S8 refined from the NPD data measured with wavelength λ = 1.15 Å using a structure model with the space group P2/c (see the text). Curly brackets and square brackets denote cation sites in the vacancy and fully occupied layers, respectively. Agreement values [33] and stoichiometry estimated from the Rietveld refinement are given at the bottom.
| Atom (Wyckoff site) | x(δx) | y(δy) | z(δz) | Occupancy (δN) |
|---|---|---|---|---|
| [Fe (4g)] | 0.2635(5) | 0.2368(11) | 0.9997(3) | 0.766(3) |
| [Ti (4g)] | 0.233(3) | |||
| {Ti (2 f)} | 1/2 | 0.2407(42) | ¼ | 0.254(5) |
| {Fe (2e)} | 0 | 0.8592(24) | ¼ | 0.225(1) |
| {Ti (2e)} | 0.276(1) | |||
| S (4g) | 0.7064(11) | 0.2478(29) | 0.1207(4) | 1 |
| S (4g) | 0.2110(11) | 0.2534(33) | 0.6279(4) | 1 |
| Unit cell: a = 5.9749(2) Å, b = 3.4407(1) Å, c = 13.0028(4) Å, β = 117.150(4)0 | ||||
| χ2 = 1.48, RB = 5.7%, Rwp =3.9%, Rexp = 3.2%; total stoichiometry Fe3.96Ti3.05S8 | ||||
As to the Co containing samples, the NPD pattern for Fe4Co3S8 can be well indexed by the 3C crystal structure model which was used for x-ray analysis and reported in [34] for Fe7Se8. In order to find the Co and Fe distributions over cation layers in this trial model we suggested that each of the five cation Wyckoff sites are equally occupied by Co and Fe atoms giving Fe3.5Co3.5S8 stoichiometry.
Then the unit cell parameters, atomic coordinates, Co and Fe occupation factors and profile function parameters were allowed to be refined. The best fit result is shown in figure 11, and refined structural parameters are represented in table 2. As can be seen from table 2, the Co and Fe atoms almost equally occupy the non-full layers (Co—37.9%, Fe—37.1%, vacancies—25%), while in the layers without vacancies, the Co : Fe ratio was estimated as 37.8% : 62.2%. Thus, a non-random distribution of Co for Fe atoms in alternating cation layers takes place in Fe4Co3S8 as well.
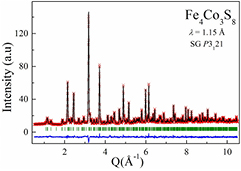
Figure 11. The best fit result on the NPD pattern measured on a Fe4Co3S8 sample with neutron wavelength λ = 1.15 Å. Symbols are experimental values of the intensity and solid lines represent the results of the Rietveld refinement using the P3121 space group. The difference between calculated and observed profiles is shown at the bottom.
Download figure:
Standard image High-resolution imageTable 2. Structural parameters of Fe4Co3S8 refined from the neutron diffraction data measured with wavelength λ = 1.15 Å using 3 C structure model with the space group P3121 (see the text). Curly brackets and square brackets denote cation sites in the vacancy and fully occupied layers, respectively. Agreement factors [33] and stoichiometry estimated from the Rietveld refinement are given at the bottom.
| Atom (Wyckoff site) | x(δx) | y(δy) | z(δz) | Occupancy (δN) |
|---|---|---|---|---|
| {Fe (6c)} | 0.0141(11) | 0.5124(9) | 0.3378(2) | 0.534(8) |
| {Co (6c)} | 0.466(8) | |||
| [Fe (6c)] | 0.5416(8) | 0.0116(9) | 0.1779(2) | 0.557(9) |
| [Co (6c)] | 0.443(9) | |||
| [Fe (3b)] | 0.0285(9) | 0 | 5/6 | 0.373(9) |
| [Co (3b)] | 0.127(9) | |||
| [Fe (3b)] | 0.4645(8) | 0 | 5/6 | 0.314(5) |
| [Co (3b)] | 0.186(5) | |||
| {Fe (3a)} | 0.0306(13) | 0 | 1/3 | 0.207(7) |
| {Co (3a)} | 0.293(7) | |||
| S (6c) | 0.1501(14) | 0.3213(18) | 0.0692(3) | 1 |
| S (6c) | 0.1782(19) | 0.3398(27) | 0.4158(4) | 1 |
| S (6c) | 0.3359(32) | 0.1603(18) | 0.2459(4) | 1 |
| S (6c) | 0.6665(24) | 0.3242(18) | 0.0872(6) | 1 |
| Unit cell: a = 6.7672(1) Å, c = 16.0795(2) Å | ||||
| χ2= 1.73, RB= 4.9%, Rwp =3.4%, Rexp = 2.6%; total stoichiometry: Fe3.95Co3.05S8 | ||||
4. Discussion and conclusions
The results of the x-ray and neutron powder diffraction and magnetization measurements performed for the Fe7−y My X8 systems (M = Ti, Co; X = S, Se) have shown that the substitution of Ti or Co for iron in both the sulfide and selenide series influences the crystal structure of compounds and leads to substantial changes in their magnetic properties. The Ti-substituted compounds are found to be in single phase state only at y ⩽ 4 and y ⩽ 3 for sulfide and selenide series, respectively, while the Co-substituted Fe7−yCoyX8 samples were determined to be single phase in the whole concentration range y = 0 – 7. The limited Ti solubility in Fe7−yTiyX8 may be associated with several factors: (i) with the absence of a stable binary Ti–S phase with the 7 : 8 stoichiometry and with NiAs-type structure (see [35], for instance); (ii) a higher valence of Ti than Fe ions, and (iii) an appreciable difference in electronegativities of titanium and iron. Despite the formation of different superstructures, all the synthesized compounds Fe7−yTiyX8 and Fe7−yCoyX8 exhibit a layered crystal structure of the NiAs-type. It has been observed, that the interatomic distances within layers remain almost unchanged upon substitutions, while the variations of the average interlayer distances are substantially different in the Fe7−yTiyX8 and Fe7−yCoyX8 systems. In Fe7−yTiyX8 compounds, the Ti for Fe substitution results in an expansion of the crystal lattice perpendicular to the layers unlike the substitution of Co for Fe in Fe7−yCoyX8 systems where the growth of the Co concentration is accompanied by an appreciable reduction of the average interlayer distances. These changes in the crystal lattice of Fe7−yTiyX8 with substitutions may be attributed to the difference in sizes of Ti and Co ions in respect to Fe. As was mentioned above, the interlayer distance in the Fe7 X8-type compounds was suggested as a key factor that governs the magnetic state of 3d ions [23, 36]. However, as follows from figure 12(a) which displays the normalized Neel temperatures as a function of the concentration of substituting atoms, the TN(y)/TN(0) dependencies for all four Fe7−yMyX8 (M = Ti, Co; X = S, Se) series behave in the identical way upon substitution despite the large difference in changes of the crystal lattice. The monotonous reduction of the magnetic ordering temperature in Fe7−yMyX8 compounds with increasing M content allows us to suggest that both the Ti and Co atoms have nearly zero magnetic moments and can be considered as diluents for the magnetic Fe sublattices. The titanium chalcogenide compounds usually exhibit diamagnetic or Pauli paramagnetic properties [37], however, when a chalcogenide compound also contains other 3d metal ions with own magnetic moments, a small magnetic moment may assumingly be induced on Ti ions as well. In particular, the presence of a small magnetic moment about of (0.1–0.4) µB on titanium atoms was suggested from neutron diffraction measurements performed for Fe0.5TiSe2 [38, 39] and Fe0.5TiS2 [40]. More intriguing is the magnetic state of cobalt in Fe7−yCoyX8. The Pauli paramagnetic behavior of Co7X8 was reported to result from the 3d-band broadening [36] in comparison with Fe7 X8 compounds because of reduced by about 10% interlayer distances. However, this suggestion contradicts with the specific heat measurements which have revealed that the Co7Se8 compound exhibits a higher value of the electronic specific heat coefficient (12.33 mJ mol−1K−2) than Fe7Se8 (10.71 mJ mol−1K−2 and 9.24 mJ mol−1K−2 for the 3C and 4C superstructures, respectively) [41]. Our data show that in Fe7−yCoyX8, the Co atom exhibits a fairly low magnetic moment even being inserted instead of Fe into the crystal lattice with high interlayer distances at y < 4. This is in agreement with the NMR data reported for Fe7−yCoyX8 [25]. It is worth to mention that according to neutron diffraction the Fe magnetic moment in Fe7S8 at low temperatures is equal to 3.16 μB [18] which is less than the expected value 4 μB for Fe2+. Such a difference may be attributed to the involvement of Fe 3d electrons in the formation of Fe–S chemical bonds. The oxidation state of Fe in Fe7S8 and Fe7Se8 is still discussed in literature (see [6, 7, 42, 43]). In earlier works, from the condition of the charge neutrality and magnetization data both the Fe2+ and Fe3+ ions were suggested to exist in Fe7S8 (see [6], for overview). However, the presence of Fe3+ was not confirmed by Mössbauer spectroscopy [7]. Besides, the absence of Fe3+ in Fe7S8 was evidenced by the x-ray MCD experiments [42]. It interesting to note that at high temperatures (above the critical temperature of the order–disorder structural transition), the effective magnetic moment per Fe ion in Fe7S8 was determined to be about 5.8 μB [26] which substantially surpasses the theoretical value 4.89 μB for Fe2+ and is close to the expected value 5.91 μB for the Fe3 +. The same situation was observed for the selenide Fe7Se8 compound [23, 44]. The enhanced μeff values derived from the high-temperature susceptibility measurements may be indicative of changes in the oxidation state of Fe with increasing temperature. The substitution of Ti for Fe in Fe7S8 was observed to result in reduction of the effective magnetic moment apparently because of the hybridization of Fe 3d and Ti 3d electronic states [26]. These results together with the previously reported x-ray MCD data [42] show that a simple ionic model is not applicable for these compounds.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
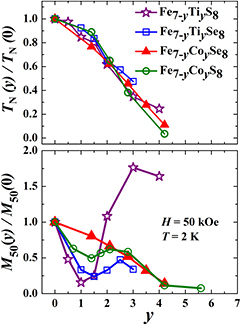
Figure 12. The relative Neel temperature TN(y)/TN(0) (upper panel) and relative magnetization M50(y)/M50(0) measured in an applied field of 50 kOe at 2 K (lower panel) as functions of the substituting atom concentration in the Fe7−yMyX8 series (M = Ti, Co; X = S, Se).
Download figure:
Standard image High-resolution image{kind=link}
The concentration dependences of the magnetization collected in figure 12 for Fe7−yMyX8 systems (M = Ti, Co; X = S, Se) clear indicate that the substitution of Ti for Fe in both the Fe7S8 and Fe7Se8 compounds and Co for Fe in Fe7S8 is non-random. The statistical distribution of substituting atoms should result in the monotonous reduction of the resultant magnetization. Such a behavior is observed in Fe7−yCoySe8. As was mentioned above, the presence of the ferrimagnetic order and resultant magnetization in Fe7X8 are associated with the absence of the compensation between two ferromagnetically ordered sublattices with antiparallel alignment of the magnetizations. If we assume that the full cation layer in Fe7X8 contains four Fe atoms and the neighbor cation layer includes three Fe atoms and one vacancy, the compensation of sublattice magnetizations in Fe7−yMyX8 can be realized by the replacement of one Fe atom in the full layer by a non-magnetic atom. Indeed, as follows from the magnetization data presented in figure 12, a pronounced minimum of the magnetization associated with the magnetic compensation is observed exactly at y = 1 in the case of the Ti for Fe substitution in the sulfide system Fe7−yTiyS8. The full compensation of the sublattice magnetizations in Fe6Ti1S8 results in the antiferromagnetic behavior of the magnetization in this compound. In the selenium system Fe7−yTiySe8, the spontaneous magnetization does not disappear upon the substitution, and the minimum of the magnetization is located at a slightly higher concentration of substituting atoms (y ≈ 1.5) apparently owing to a lower degree of partitioning. The same tendency is observed in the case of the Co substitution: the cation partitioning obviously exists in the sulfide system Fe7−yCoyS8, while in the case of the selenide Fe7−yCoySe8 compounds, the distribution of Co atoms seems to be more random.
When comparing the Ti for Fe and Co for Fe substitutions it is clear from the magnetization measurements and neutron diffraction data that the Ti-substituted Fe7−yTiyX8 systems exhibit a substantially increased degree of cation partitioning than the Co-substituted counterparts. Such a difference apparently originates in the different spatial extensions of Ti 3d and Co 3d orbitals and in abilities of the cations to delocalize electrons by direct orbital overlap. As was suggested by Goodenough [45], the overlap between cation orbitals in the 3d transition-element compounds is controlled by a critical cation–cation separation, and the overlap is more effective for cations from the earlier part of the transition-element series. From this point of view, more pronounced partitioning can be obtained when one of the two different cations in the compound belongs to the first half while another one to the second half of 3d series, like titanium and iron in our case. The minimum of the resultant magnetization observed in Fe7−yMyX8 at the M concentrations y ≈ 1 – 1.5 implies that the substituting atoms initially occupy the cation layers without vacancies, while the layers with vacancies are occupied with further growth of the M content. The last is confirmed by neutron diffraction data for Fe4Ti3S8 and Fe4Co3S8. Less pronounced partitioning of 3d ions in the adjacent cation layers in selenides Fe7−yMySe8 in comparison with sulfides apparently originates in the lesser overlap of 3d -orbitals because of the larger interatomic distances since selenium has a bigger ionic radius than sulfur. One can assume that 3d electrons are more localized in the selenide compounds Fe7−y MySe8 than in Fe7−y MyS8. It should be noted that the site-preferential occupation was revealed by neutron diffraction measurements in some other transition-metal chacogenides having the NiAs-type structures, in particular, in  M3−xS4 [46–48].
M3−xS4 [46–48].
As to the enhancement of magnetic hardness observed in Fe7−yTiyX8 compounds with increasing Ti concentration, there are several examples of the iron-containing chalcogenide compounds exhibiting extremely high coercivities at low temperatures. Thus, giant values of the coercive field (from 40 up to 90 kOe) at low temperatures were observed in Fe0.5TiS2 [40] and in Fe0.28TaS2 [49] and Fe0.25TaS2 [50] having layered crystal structures as well. The anisotropy field in FexTaS2 compounds is estimated to be above 500 kOe [49–51]. As evidenced by x-ray MCD measurements, the magnetic moment of Fe ions in these compounds exhibit a substantial orbital contribution (up to 1 μB [51]). Since an unquenched orbital moment was also detected in Fe7S8 [42], the increased magnetic hardness of Fe7−yTiyX8 compounds with high Ti concentrations (x > 2) may originate in the enhancement of orbital moment of Fe ions owing to the changes in crystal electric fields upon Ti for Fe substitutions. The Co for Fe substitution in Fe7−yMySe8 apparently has the opposite influence.
In conclusion, the impact of the Ti for Fe and Co for Fe substitutions on the crystal structure and magnetic properties of the Fe7S8 and Fe7Se8 compounds with a layered crystal structure has been systematically studied. The substitutions in the cation layers are observed to be substantially non-random especially in the case when Fe atoms are replaced by titanium. The degree of the cation partition between adjacent layers is found to depend on the atomic number of substituting 3d atoms in respect to Fe as well as on the type of chalcogen in Fe7−yMyX8 systems. The results obtained in the present work show that the layer-preferential substitutions may be used to tune the properties of Fe chalcogenides with layered crystal structures of the NiAs-type.
Acknowledgments
This work is partly based on experiments performed at the Swiss spallation neutron source SINQ, Paul Scherrer Institute, Villigen, Switzerland. This work was supported by the Ministry of Education and Science of Russia (project No 1362), by the Russian Foundation for Basic Research (projects No 13-02-00364 and 13-02-96038), by the program of UB of RAS (project No 15-17-2-22) and by the Government of Sverdlovsk region.