

Abstract
Exposure to ionising radiation is clearly associated with an increased risk of developing some types of cancer. However, the contribution of non-targeted effects to cancer development after exposure to ionising radiation is far less clear. The currently used cancer risk model by the international radiation protection community states that any increase in radiation exposure proportionately increases the risk of developing cancer. However, this stochastic cancer risk model does not take into account any contribution from non-targeted effects. Nor does it consider the possibility of a bystander mechanism in the induction of genomic instability. This paper reviews the available evidence to date for a possible role for non-targeted effects to contribute to cancer development after exposure to ionising radiation. An evolution in the understanding of the mechanisms driving non-targeted effects after exposure to ionising radiation is critical to determine the true contribution of non-targeted effects on the risk of developing cancer. Such an evolution will likely only be achievable through coordinated multidisciplinary teams combining several fields of study including: genomics, proteomics, cell biology, molecular epidemiology, and traditional epidemiology.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The relationship between non-targeted effects and the risk of developing radiation-induced cancer is still quite uncertain with a significant amount of the data being null, inconclusive, contradictory, or open to alternate interpretation. The current estimate of cancer risk (5% per sievert) is based on epidemiological studies of large populations (such as the Japanese atomic bomb survivors) with relatively high radiation doses [1, 2]. Cancer risk from low dose exposures (i.e. below 100 mGy) is extrapolated linearly down to zero for which no firm epidemiological evidence exists. This extrapolation, known as the linear-non-threshold (LNT) model, is based on the assumption that the physical energy deposited by ionising radiation (or simply radiation) increases carcinogenic risk linearly with increasing dose, regardless of dose rate [2]. This conclusion is contentious because the LNT model cannot explain all biological findings. The LNT model has been repeatedly endorsed for radiation protection purposes—that is, for setting dose limits and as the basis for ALARA (As Low As Reasonably Achievable) requirements; it is not intended for risk analysis [2–5]. Further, accumulating evidence suggests that the LNT model does not accurately reflect the mechanistic contributions of radiation effects. More refined models which take into account personal dosimetry, age, and sex are required for determining individual risk after radiation exposure. To this end, risk analysis models will have a major role to play in future studies and validation by other groups will be necessary [6, 7].
The weight of evidence provided by epidemiological studies, which has been typically used to support the LNT model, indicates that if an excess risk from low dose radiation exposures exists, it is indistinguishable from baseline cancer incidence or mortality risk. Overall, low dose exposure studies typically find cancer mortality and incidence rates that are in line with what is expected for the general population or demonstrate a healthy worker effect [2, 3, 8, 9]. In contrast, some recent studies, evaluating moderate to low dose exposures, have been able to detect a statistically significant increase in the risk for select cancers following radiation exposures below 100 mGy due to their strong statistical power, few confounding factors, and narrow confidence intervals [10–16]. The evidence of a risk below 100 mGy is becoming more frequent in the latest research. These studies are well designed and thus may offer greater insights into low dose risk that were previously not detected. They highlight the need for further investigation.
Cancers are a large group of diseases that result in cells becoming defective in normal metabolic and cell proliferation pathways. Tumour development can be divided into three phases: tumour initiation, tumour promotion, and malignant progression. Tumours frequently initiate following molecular damage to DNA [17]. Damage often involves activation of growth promoting genes (oncogenes) or disruption of growth inhibitory signals (tumour suppressors). The tumour promotion stage is the accumulation and amplification of the initial damage by cellular responses in an attempt to maintain the genetic stability of the cell [18]. The progression stage occurs when cells continue to proliferate as a result of the acquisition of additional alterations [19]. The accumulation of mutations in up to 20 different genes is required to overcome the multiple safeguards that exist to protect against the development of an invasive cancer. The majority of these mutations confer only a small selective advantage during tumorigenesis [20]. Thus, it is best to think of mutations as contributing to (rather than causing) cancer [21]. Several studies have attempted to characterise exposure to radiation as a direct initiator, promoter, or progressor of cancer [22–27]. However, it is difficult to strictly classify radiation as one or the other because it could affect carcinogenesis as an initiator, promoter, or progressor, depending on dose, the receptor [28, 29], and potentially as a function of age at exposure [6]. For this reason, particular attention is paid in this review to the dose, the receptor, and the general state of the target cell.
It has been known for quite some time that un-irradiated cells can and do respond to radiation-induced damage. These responses are collectively known as non-targeted effects and are well documented, even though the underlying mechanisms are not completely understood. Two main types of non-targeted effects include the bystander effect and genomic instability. Radiation-induced bystander effects refer to the presence of radiation-like effects in cells that were not directly hit by radiation, and involve cellular signalling pathways [5]. Radiation-induced genomic instability refers to the increased acquisition rate of alterations or mutations in the genome being transmitted to progeny cells [5]. These cellular and genomic responses have been put forward as evidence that the response to radiation-induced DNA damage may, in some cases, deviate from linearity. However, at this time, it is not certain whether non-targeted effects are causing a deviation from linearity, nor if the deviation is leading to an overestimation or an underestimation of cancer risk.
2. Methods
The purpose of this comprehensive review is to summarise and critically analyze the relevant scientific research on the role of non-targeted effects in cancer development in hopes of identifying gaps or inconsistencies to generate future research avenues. Non-targeted effects are important not only for high radiation dose exposures, but low dose exposures as well. Furthermore, there is limited knowledge available on the interaction of non-targeted effects such as the bystander effect and genomic instability. This topic was originally chosen as the focus of a graduate review project to provide the Canadian Nuclear Safety Commission with a scientifically sound knowledge base and several options for which to provide funding for future research. Pubmed was the primary electronic database used to search for relevant literature. By using the advanced search option, the keyword search terms selected (all fields) were: radiation, bystander effect, genomic instability, and cancer. From the approximately 70 papers identified, additional papers were identified based on the citations of the original 70 papers. This included several books and international reports. The method by which papers were included or excluded from this work was based on a set of detailed questions which addressed the adequacy of the experimental design, data analysis, consideration of alternate explanation of findings, biological plausibility of findings, and sufficient evidence to support conclusions.
3. Review results
3.1. Historical experimental approach
In order to study non-targeted effects, researchers have used a microarray approach to see how radiation dose can impact gene expression. These studies have used high dose, low dose, and shielded dose components and statistically significant changes in gene expression can be seen for each category. Gene expression patterns depend on the cell type, the growth state of the cell, the type of radiation, and the biological endpoint being measured. The types of genes expressed following exposures to doses below about 200 mGy and scatter doses vary substantially from those expressed in response to higher doses and there may also be important time- and tissue-dependent differences [30–40]. Ding et al [41] noted that the genes upregulated in response to high dose radiation exposures tend to be involved in regulating apoptosis and cell proliferation, while the genes upregulated in response to low dose radiation exposures tend to be involved in signal transduction, intercellular signalling, and DNA damage responses. Although a coherent set of genes that responds differently to high and low doses of radiation has yet to be identified [42], many of these genes have been implicated in cancer development. Select genes and signalling pathways are highlighted in the following sections as they may provide a link between radiation exposures and increased cancer risk via bystander effects, activation of chronic inflammation, and stimulation of genomic instability events.
3.2. Radiation-induced bystander effect
Cell–cell communication is required to mediate the bystander effect and is thought to involve two main mechanisms: the secretion of soluble factors into the extracellular space [43–45]; and via cell–cell interchanges through gap-junction communication [46, 47]. There is now firm evidence that this phenomenon does not only occur in vitro, but in vivo as well [48, 49].
A variety of end points have been reported as bystander effects, such as sister chromatid exchanges [50–54], the presence of chromosomal rearrangements and aberrations [55, 56], mirconuclei formation [47, 57–59], the induction of gene mutations including acquisition of point mutations (such as base changes) [60–66], gene expression changes [57, 67] including changes in γH2AX foci that indicate DNA damage [67–69], changes in cell proliferation, cell cycle control, cell transformation, cell death, or apoptosis [56, 70–79]. Radiation-induced bystander effects have been proposed to require the involvement of several biological signalling pathways, including both p53-independent [80] and p53-dependent pathways [47, 81] as well as ATM-dependent pathways [54].
The evolution in our understanding of the potential mechanisms behind the bystander effect comes directly from gene expression studies. Huo et al [66] confirmed that the types of mutations occurring in bystander cells were primarily point mutations, whereas those occurring in irradiated cells were primarily deletions. Since reactive oxygen species (ROS) are thought to induce primarily point mutations, they propose that generation of ROS is a major mechanism by which mutations arise in bystander cells [66]. Furthermore, bystander and directly irradiated cells respond to radiation differently with regard to cell signalling [82]. It is not only important to understand the mechanism behind the bystander effect, but also what, if any, role it may play in radiation-induced carcinogenesis. Watson et al [83] provided the first evidence for an in vivo bystander mechanism in the induction of chromosomal instability in response to ionising radiation. Several papers support these findings [84–88]. Table 1 illustrates how inflammatory mediators link the bystander effect and genomic instability.
Table 1. Dose and observed bystander effect.
| Reference | Dose | Target tissue | Response of target | Effect |
|---|---|---|---|---|
| Mancuso et al [46] | 3000 mGy (direct and indirect) (x-rays) | Patched-1 heterozygous mouse cerebellum | Direct: Whole body irradiated animals developed aggressive cerebellar tumours. | An increased tumour response in shielded tissue suggests that a persisting low level of DNA damage in central nervous system after low radiation doses (36 mGy, estimated scatter dose) may facilitate chromosomal events relevant for carcinogenesis. |
| Indirect: Animals with shielded brains had a lower tumour response than directly irradiated animals but significantly more tumours than untreated animals. | ||||
| • In vivo evidence of a dose-response effect. | ||||
| • Bystander effects are in vivo events with carcinogenic potential | ||||
| Lorimore et al [85] | 4000 mGy (direct and indirect) (gamma radiation) | Whole body mice (CBA/Ca and C57BL/6) | Macrophages obtained from the bone marrow of irradiated CBA/Ca, but not C57BL/6 mice, are able to induce chromosomal instability assayed as nonclonal cytogenic aberrations in the clonal descendants of non-irradiated stem cells | Chromosomal instability was induced as a consequence of proinflammatory cytokine signalling. The findings show a genotype dependent chromosomal instability phenotype induced by radiation-induced macrophage-mediated bystander signalling in vivo. |
| • TNF-α was implicated in the mechanism underlying the macrophage-mediated chromosomal instability phenotype. | ||||
| Ghandhi et al [82] | 500 mGy (direct and indirect) (alpha particles) | Primary human lung fibroblasts | Direct: P53- and NFκB-regulated radiation response genes were expressed at elevated levels in the directly exposed cells. | Two major transcriptional hubs (p53 and NFκB) that regulate the response to radiation responded differently in bystander cells and similarly in directly irradiated cells in vitro. |
| Indirect: P53-regulated radiation response genes (ex: CDKN1A) showed little or no change in bystander cells. Genes regulated by NFκB (ex: COX-2) were expressed at elevated levels in bystander cells. | ||||
| • 'The greater relative contribution of signalling through NFκB in bystander cells may lead to a greater survival of these cells, even in the presence of persistent damage, possibly putting bystander cells at an increased risk for long-term consequences of radiation damage.' | ||||
| Chai et al [49] | 5000 mGy (direct and indirect) (x-rays) | Lower abdominal area of gpt delta transgenic mice | • No significant change in cytokine plasma levels | The TGFβ-TGFBR1-COX-2 signalling pathway has a critical role in radiation-induced non-targeted response and genomic instability in vivo. |
| • Significant change in cytokine levels in non-targeted lung tissues, but not in non-targeted liver tissues | ||||
| • COX-2 signalling pathway falls under NFκB regulation |
3.3. Radiation-induced genomic instability
Radiation-induced genomic instability is characterised by the accumulation of genetic alterations within individual cells that increases over time and is transmitted to progeny cells, even multiple generations after the initial insult [89, 90]. The progeny of the irradiated cells display a high level of non-clonal genomic damage, which is present at a frequency that is not consistent with independent mutations [91]. The induction of radiation-induced genomic instability appears to be dependent on radiation quality (high- or low-linear energy transfer (LET)) and genetic background [92, 93]. There may also be a threshold for low-LET radiation (<500 mGy), below which no genomic instability is induced [91, 94].
Some of the measured endpoints for genomic instability overlap with the end points mentioned above for the bystander effect, demonstrating the commonalities between the two radiation-induced non-targeted effects. The endpoints used to characterise genomic instability have included: increased levels of sister chromatid exchanges [40, 45, 95] and chromosomal breaks or aberrations [81, 91, 96–98]; enhanced micronuclei formation, gene mutations and amplifications [96, 99]; changes in ploidy [100–102]; mini- and micro-satellite instabilities and/or decreased plating efficiency [90, 103]; increased cellular transformation, and enhanced rates of apoptosis [104]; and changes in the incidence of cell death [66, 105, 106].
The mechanisms by which exposure to radiation results in the induction of genomic instability are still unclear [107]. Several different mechanisms have been proposed, including: bridge-breakage; persistent oxidative stress affecting DNA regulation; unequal DNA segregation; and, altered DNA regulation [96, 108, 109]. However, two mechanisms that are gaining support are that of epigenetics [110] and chronic inflammation [111]. Epigenetics refers to heritable changes in gene expression that take place without alterations in the DNA sequence. Epigenetic changes include an array of molecular modifications such as DNA methylation, histone acetylation, chromatin remodeling, and miRNA regulated gene expression [112–116]. Chronic inflammation refers to activation of innate immune processes that result in downregulation of DNA repair pathways and cell cycle checkpoints in response to release of inflammatory mediators and ROS which lead to genomic instability [111]. The mechanisms leading to genetic alterations under inflammatory conditions are still unclear and have been the focus of many recent reviews [111, 117–119].
3.4. Implications for non-targeted radiation-induced carcinogenesis
As stated above, Watson et al [83] provided the first evidence of an in vivo bystander mechanism in the induction of chromosomal instability in response to ionising radiation. Furthermore, inflammatory processes provide a mechanistic link between radiation-induced chromosomal instability, bystander effects, and clastogenic factors [85]. Many studies have demonstrated that the nuclear factor (NF)-κB family of transcription factors plays an important role in radiation-induced bystander effects [120] as well as immune/inflammatory responses [119]. Chai et al [49] have shown that the TGFβ-TGFBR1-COX-2 signalling pathway plays a critical role in the radiation-induced non-targeted response in vivo. The COX-2 signalling pathway is under NFκB regulation [120] and is over-expressed in tissues subjected to chronic inflammation and infiltrating leukocytes [121]. Also, there is evidence that TGFβ is involved in ROS induction and Ca2+ influx by bystander cells [122, 123]. Various links between the bystander effect, chronic inflammation, and genomic instability exist. Additionally, there is recent evidence that examining different epigenetic responses in bystander organs after partial body irradiation may provide further insight into the mechanism leading to genomic instability [124].
Until recently, the bystander effect, chronic inflammation, and genomic instability were typically studied separately. By combining all of these radiation-related events, a more global model of non-targeted radiation carcinogenesis can be established (see figure 1). Figure 1 shows a cell being directly hit by ionising radiation in frame (a), followed by a radiation-induced bystander event in frame (b). ROS are proposed to be the mechanism by which mutations arise in bystander cells. Inflammatory mediators decrease DNA repair pathways and cell cycle checkpoints during bystander cell division (frame c). Additional signalling from inflammatory mediators leads to an instability event in the progeny of the bystander cell in frame (d). Epigenetic regulation is proposed to be the means by which multiple non-targeted events facilitate carcinogenesis.
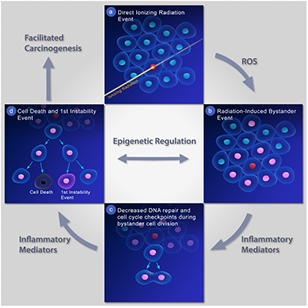
Figure 1. Proposed mechanistic model of non-targeted radiation-induced carcinogenesis.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusions
Although there is no strong epidemiological evidence of excess cancers caused directly by non-targeted effects following radiation exposure, the studies reviewed here provide a mechanistic link between the bystander effect and genomic instability in the induction of radiation-induced carcinogenesis. As limited (experimental) human data exists, the animal studies reviewed here can inform radiation protection practices for people exposed to radiation from occupational, medical, and natural sources. Animal studies have shown that bystander effects can increase the tumour response in shielded mice [46], are genotype dependent [85], and can induce multiple signalling pathways [49]—supporting the findings that bystander cells could be at a greater risk than irradiated cells for long-term consequences of radiation damage [82]. The evidence presented in this review does not warrant a deviation from current radiation protection practice, but it does show that additional work is required to determine the extent to which non-targeted effects contribute to radiation-induced carcinogenesis. Future studies will require coordinated multidisciplinary teams combining several fields of study including: genomics, proteomics, cell biology, molecular epidemiology, and traditional epidemiology to achieve a greater understanding of the contribution of non-targeted effects in radiation-induced carcinogenesis.
Acknowledgments
The authors wish to acknowledge the assistance of Annick Laporte in the graphic design of figure 1 and to thank the two anonymous reviewers for their valuable suggestions.