


ABSTRACT
In an effort to provide an accurate spectroscopic characterization of oxirane, state-of-the-art computational methods and approaches have been employed to determine highly accurate fundamental vibrational frequencies and rotational parameters. Available experimental data were used to assess the reliability of our computations, and an accuracy on average of 10 cm−1 for fundamental transitions as well as overtones and combination bands has been pointed out. Moving to rotational spectroscopy, relative discrepancies of 0.1%, 2%–3%, and 3%–4% were observed for rotational, quartic, and sextic centrifugal-distortion constants, respectively. We are therefore confident that the highly accurate spectroscopic data provided herein can be useful for identification of oxirane in Titan's atmosphere and the assignment of unidentified infrared bands. Since oxirane was already observed in the interstellar medium and some astronomical objects are characterized by very high D/H ratios, we also considered the accurate determination of the spectroscopic parameters for the mono-deuterated species, oxirane-d1. For the latter, an empirical scaling procedure allowed us to improve our computed data and to provide predictions for rotational transitions with a relative accuracy of about 0.02% (i.e., an uncertainty of about 40 MHz for a transition lying at 200 GHz).
Export citation and abstract BibTeX RIS
1. INTRODUCTION
Our knowledge on the universe chemical inventory has been obtained and continuously updated by means of astronomical observations (Tielens 2013), which have revealed a plethora of molecular species, thus demonstrating the rich chemistry of the universe. The observation of gas-phase species is usually performed using rotational spectroscopy, which can be undertaken from the ground (Herbst & van Dishoeck 2009), while vibrational spectroscopy observations, for which ground-based observations are hampered by the terrestrial atmosphere, are mainly carried out by spectrometers on board satellites or airbornes. In particular, infrared (IR) spectroscopy is suitable for retrieving the chemical composition of planetary atmospheres. To give an example of interest in the context of this paper, the atmosphere of Titan, the largest moon of Saturn and the second largest moon in our solar system, has been fully characterized by recording its IR spectrum (200–1500 cm−1) with the infrared radiometer spectrometer on board the Voyager 1 mission first, with the Infrared Space Observatory (ISO), in orbit around Earth in 1997, and more recently with the instruments on board the Cassini spacecraft.
The great interest in Titan's atmosphere is due to the fact that recent measurements (Niemann et al. 2002; Waite et al. 2004; Young et al. 2004) revealed that Titan's atmosphere is characterized by a rich and complex organic chemistry (see Ali et al. 2013 and references therein). This led to a further interest in Titan because the investigation of its atmosphere might shed light on the organic evolution in the atmosphere of early earth. In fact, Titan is thought to represent a model of primitive earth (Raulin et al. 2009) because its atmosphere contains significant quantities of carbon (∼2% CH4) and nitrogen (98% N2), and traces of oxygen (∼50 ppm CO). Photochemical production of complex molecules containing C, N, O, and H is therefore possible, and consequently, the investigation of Titan's atmosphere might provide an unique opportunity to explain the terrestrial prebiotic chemistry.
The presence of organic molecules in Titan's atmosphere was revealed by IR spectra observations from Voyager 1 (Hanel et al. 1981; Kunde et al. 1981), but it was with the Cassini–Huygens mission that the chemical complexity of Titan's atmosphere was fully exploited. Following the discovery of CO, CO2, and more recently of H2O, the possible generation of oxygenated organic compounds was theoretically and experimentally investigated. In particular, Coll et al. (2003) and Bernard et al. (2003) carried out experimental simulations of Titan's atmosphere. By using cold plasma flow reactors and mixtures of N2/CH4/CO (98/2/0.01), they investigated the possible formation of O-bearing compounds in order to determine which minor species could be present in Titan's atmosphere. Surprisingly, they identified the presence of oxirane (also named ethylene oxide, c-C2H4O), which was initially unpredicted, even if it was already observed in the interstellar medium (Dickens et al. 1997; Ikeda et al. 2001; Turner & Apponi 2001). As mentioned above, the chemistry of Titan's atmosphere might provide hints on the formation of terrestrial prebiotic compounds. In fact, Cassini Ion Neutral Mass Spectrometer measurements pointed out the presence of large molecules (>100 u), thus heightening astrobiological interest in this satellite. This interest led to several investigations. Among these investigations, we mention the work of Hörst et al. (2012): by experimentally simulating the aerosols produced in Titan's atmosphere, the authors demonstrated that biological molecules can be produced. In particular, the presence of cytosine, uracil, thymine, guanine, glycine, and alanine was confirmed by analyses with high-resolution mass spectrometry and gas chromatography–mass spectrometry.
While suggesting that small carbonaceous molecules may be responsible for unidentified infrared bands (UIRs), Bernstein & Lynch (2009) demonstrated a close correlation between the IR spectra of oxirane and cyclopropenylidene (c-C3H2) and the major UIR bands. However, considerable uncertainties in some of the assignments still persist and the authors pointed out the need of quantitative simulations of UIR spectra that properly account for anharmonicity effects on both line positions and intensities. Despite the fact that the IR absorption spectrum of oxirane has been investigated both experimentally and theoretically (Bennett et al. 2005; Nakanaga 1980, 1981; Yoshimizu et al. 1975; Lord & Nolin 1956; Bertie & Othen 1973; Nyquist & Putzig 1986; Russell & Wesendrup 2003; Johnson et al. 2004; Lafferty et al. 2013; Flaud et al. 2012; Kwabia Tchana et al. 2013, 2014), there are still uncertainties in the assignment of some spectral features because of their weakness and/or because they are hidden by stronger, overlapping nearby bands. Furthermore, several bands were assigned based on the liquid Raman or solid state spectra, thus introducing some uncertainty due to condensed phase interactions.
In view of the astrochemical relevance of oxirane and the issues still open on its presence in different astronomical objects, with particular emphasis on Titan's atmosphere, we decided to carry out a thorough spectroscopic characterization by means of state-of-the-art computational methods and approaches. Particular attention is paid to the proper assignment of its IR spectrum. Since very high D/H ratios, i.e., up to 1000 times that of the Galactic value, have been determined for some interstellar molecules (see, for example, Ceccarelli 2002; Roueff & Gerin 2003), the accurate determination of the rotational parameters and IR spectra for the mono-deuterated species, oxirane-d1, has also been considered.
2. COMPUTATIONAL DETAILS
The coupled-cluster (CC) singles and doubles approximation augmented by a perturbative treatment of triple excitations [CCSD(T)] (Raghavachari et al. 1989) was employed in molecular structure and force-field calculations in conjunction with the correlation-consistent basis sets, (aug)-cc-p(C)VnZ (n = T,Q,5) (Dunning 1989; Kendall et al. 1992; Woon & Dunning 1995). CCSD(T) calculations were carried out with the quantum-chemical CFour program package.4
To accurately determine the equilibrium structure of oxirane, a composite scheme was employed. This approach is based on additivity at an energy-gradient level (Heckert et al. 2005, 2006), with the energy gradient used in the geometry optimization being

In this expression, dE∞(HF-SCF)/dx and dΔE∞(CCSD(T))/dx are the energy gradients corresponding to the exp(− Cn) expression for the extrapolation to the complete basis set (CBS) limit for Hartree–Fock Self-consistent field (HF-SCF; Feller 1993) and to the n−3 extrapolation formula for the extrapolation of the CCSD(T) correlation contribution (Helgaker et al. 1997), respectively. In the formula given above, n = T, Q and 5 were chosen for the HF-SCF extrapolation, while n = Q and 5 were considered for CCSD(T). Core-valence correlation effects were included by adding the corresponding correction, dΔE(CV)/dx, with the core-correlation energy correction, ΔE(CV), being obtained as the difference between all-electron and frozen-core CCSD(T) energies using the core-valence cc-pCVQZ basis set.
The spectroscopic investigation is based on harmonic and anharmonic force-field determinations at the CCSD(T)/(aug)-cc-p(C)VnZ levels (with n = T,Q). As implemented in CFour, the harmonic part was obtained using analytic-second derivatives of the energy (Stanton & Gauss 1997), whereas the corresponding cubic and semi-diagonal quartic force constants were obtained in a normal-coordinate representation via numerical differentiation of the analytically evaluated force constants (Stanton et al. 1998; Stanton & Gauss 2000; Thiel et al. 1998; Schneider & Thiel 1989). The harmonic force field was improved by means of a composite scheme aimed at accounting for basis-set truncation and core correlation, thus deriving a best-estimated harmonic force field. Following the suggestions by Tew et al. (2007), the harmonic frequencies ω were extrapolated to the CBS limit (ω(CBS)) starting from the results obtained at the CCSD(T)/cc-pVTZ and CCSD(T)/cc-pVQZ levels. More precisely, the extrapolated correlation contribution was added to the HF-SCF CBS limit, which was assumed to be reached at the HF/cc-pV5Z level. Corrections due to core correlation were then evaluated at the CCSD(T)/cc-pCVTZ level (Δω(CV) = ω(CCSD(T)/CVTZ, all) − ω(CCSD(T)/CVTZ, fc)). According to our previous work (see, for example, Barone et al. 2013; Pietropolli Charmet et al. 2013), the effects of diffuse functions in the basis set can play a role. For this reason, the harmonic force field was also evaluated at the CCSD(T)/aug-cc-pVTZ level and the corresponding correction (Δω(diff) = ω(CCSD(T)/augVTZ, fc) − ω(CCSD(T)/VTZ, fc)) included as follows:

The computations of vibrational spectra beyond the double-harmonic approximation was performed by making use of second-order vibrational perturbation theory (VPT2) (Nielsen 1951; Mills 1972; Isaacson et al. 1981; Amos et al. 1991) applied to the hybrid force field generated by replacing the harmonic part of the CCSD(T)/cc-pVnZ (n = T,Q) anharmonic force field with the best-estimated harmonic frequencies (the corresponding force fields are denoted as CC(Best)/CC(VnZ)). Such a hybrid approach is well documented in the literature (see, for example, Puzzarini et al. 2010; Pietropolli Charmet et al. 2013; Barone et al. 2014 and references therein) and allows to improve both line position and line intensity determination. VPT2 is known to be a cost-effective approach, but it requires to overcome problems related to singularities, known as resonances, plaguing the perturbative treatment. Therefore, we resorted to the so-called generalized second-order perturbation theory (GVPT2) model (Amos et al. 1991; Barone 1994, 2005). Within the latter, the resonant terms (due to the so-called Fermi resonances) were identified by means of semi-empirical criteria based on Martin's test (Martin et al. 1995) and removed from the VPT2 treatment. Subsequently, the removed terms were treated by means of a proper reduced-dimensionality variational approach. Concerning the computation of IR intensities, we resorted to the recently developed general VPT2 approach to compute transition intensities for fundamentals, overtones and combination bands by Bloino & Barone (2012). Contrary to the GVPT2 model used for line positions, intensities were computed within the deperturbed model DVPT2, which does not consider any variational treatment of the resonant terms. These VPT2 computations were performed employing a locally modified version of the Gaussian (Frisch et al. 2009) suite of programs for quantum chemistry.
Moving to the field of rotational spectroscopy, best-estimated ground-state rotational constants,  , were obtained by adding the vibrational corrections at the CCSD(T)/cc-pVQZ level to the equilibrium rotational constants,
, were obtained by adding the vibrational corrections at the CCSD(T)/cc-pVQZ level to the equilibrium rotational constants,  , corresponding to the best-estimate equilibrium structure, as follows (Mills 1972):
, corresponding to the best-estimate equilibrium structure, as follows (Mills 1972):
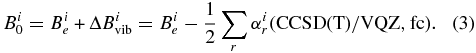
In Equation (3), the sum is taken over all fundamental vibrational modes r and i refers to the inertial axes (a, b, and c). In addition, we also considered the electronic contribution, which is related to the rotational g tensor via the following relationship (Flygare 1974):

where me and mp are the mass of the electron and proton, respectively. The rotational g tensor calculations were performed at the CCSD(T)/aug-cc-pVQZ level using our best geometries together with perturbation-dependent basis functions (Gauss et al. 1996; for more details we refer to Puzzarini et al. 2008, 2010).
By making use of the force fields computed at different levels of theory, best estimates for the quartic and sextic centrifugal-distortion constants were derived by using a composite scheme analogous to that employed for harmonic frequencies, but without accounting for the effects of diffuse functions in the case of sextic centrifugal-distortion terms. The vibration–rotation interaction constants as well as the quartic and sextic centrifugal-distortion constants were determined by means of a VPT2 treatment, as implemented in the CFour program package. The recent implementation of sextic centrifugal-distortion constants in CFour is described in Puzzarini et al. (2012).
3. RESULTS
3.1. Rotational Spectroscopy
The computed vibrational ground-state rotational and centrifugal-distortion constants of oxirane and its mono-deuterated isotopologue are collected in Table 1, where they are compared with the available experimental data. In passing, we note that the Watson's A reduction in the Ir representation (Watson 1977) is considered. For the main isotopic species, a very good agreement between our best estimates and experiment is evident, with discrepancies on the order of 0.1%, 2%–3%, and 3%–4% for rotational, quartic, and sextic centrifugal-distortion constants, respectively. We furthermore note that when the CCSD(T)/cc-pVTZ level of theory is employed for the evaluation of vibration–rotation interaction and centrifugal-distortion constants, the discrepancies negligibly change for rotational and quartic centrifugal-distortion constants (with only the exceptions of DJK) and increase (with two exception, ϕJ and ϕK, that show larger deviations) to about 4%–5% for sextic centrifugal-distortion constants. By inspection of our quantum-chemical results, it is evident why the ΦJ and ϕJ could not be experimentally determined; in fact, it turns out that they are at least three orders of magnitude smaller than the other sextic centrifugal-distortion constants. In passing, we note that ϕJ could be eventually determined by extending the rotational spectrum investigation in the far-infrared (FIR) region (Medcraft et al. 2012).
Table 1. Ground-state Spectroscopic Parameters of Oxirane and Oxirane-d1
| Parameter | Oxirane | Oxirane-d1 | |||||
|---|---|---|---|---|---|---|---|
| CCSD(T)/ | Best | Experimentc | CCSD(T)/ | Scaledd | Experimente | ||
| cc-pVTZa | Estimateb | cc-pVTZa | |||||
| A | (MHz) | 25528.576 | 25528.216 | 25483.86060(96) | 24292.883 | 24250.3320 | 24252.47(17) |
| B | (MHz) | 22122.424 | 22121.162 | 22120.87122(88) | 19907.832 | 19906.4347 | 19905.34(17) |
| C | (MHz) | 14111.338 | 14110.762 | 14097.82440(55) | 13339.993 | 13327.2181 | 13327.40(17) |
| ΔJ | (kHz) | 20.208 | 20.537 | 20.68572(77) | 15.327 | 15.689 | |
| ΔJK | (kHz) | 18.379 | 21.630 | 20.91000(216) | 21.068 | 22.401 | |
| ΔK | (kHz) | 28.123 | 26.725 | 27.59143(275) | 16.980 | 17.458 | |
| δJ | (kHz) | 6.089 | 6.104 | 6.210757(211) | 4.043 | 4.124 | |
| δK | (kHz) | 18.300 | 17.654 | 18.10778(120) | 15.322 | 16.214 | |
| ΦJ | (Hz) | −0.0007 | −0.0009 | 0.0054 | 0.0060f | ||
| ΦJK | (Hz) | 0.5805 | 0.5892 | 0.61135(302) | 0.1617 | 0.1703 | |
| ΦKJ | (Hz) | −2.4802 | −2.5198 | −2.5999(68) | −0.8288 | −0.8688 | |
| ΦK | (Hz) | 2.1118 | 2.1496 | 2.2142(50) | 0.7224 | 0.7575 | |
| ϕJ | (Hz) | −0.0011 | −0.0012 | −0.001324(179) | 0.0019 | 0.0022 | |
| ϕJK | (Hz) | 0.2627 | 0.2666 | 0.27716(153) | 0.0768 | 0.0811 | |
| ϕK | (Hz) | −0.2704 | −0.2769 | −0.3297(43) | 0.0506 | 0.0617 | |
Notes. aVibration–rotation interaction, quartic and sextic centrifugal-distortion constants computed at the CCSD(T)/cc-pVTZ level. Equilibrium rotational constants corresponding to the best-estimated equilibrium structure. Rotational constants also account for electronic corrections at the CCSD(T)/aug-cc-pVQZ level. See text. bVibration–rotation interaction at the CCSD(T)/cc-pVQZ level and electronic corrections at the CCSD(T)/aug-cc-pVQZ level. Equilibrium rotational constants corresponding to the best-estimated equilibrium structure. Best-estimated quartic and sextic centrifugal-distortion constants. See text. cExperiment: Medcraft et al. (2012). dScaled spectroscopic parameters. See text. eExperiment: Hirose (1974). fDue to the missing experimental datum, value scaled using the theoretical best estimate for the main isotopologue.
Download table as: ASCIITypeset image
For the main isotopic species, our best-estimated parameters lead to the prediction of rotational transitions with a relative accuracy of 0.1% in the centimeter/millimeter-wave region, which increases up to 0.15% in the FIR. This means that our computed parameters can predict rotational frequencies of 100 GHz and 2 THz with an accuracy of 100 MHz and 2 GHz, respectively. Moving to mono-deuterated oxirane, for which the rotational spectrum has been investigated only in the 10–60 GHz frequency range (Hirose 1974), the predictive capabilities of our computed constants were further improved by means of an empirical scaling procedure. For a generic spectroscopic parameter X, the scaling is performed using the following expression:

where the superscripts iso and main refer to the isotopic substituted species and main isotopologue, respectively. scal, exp, and calc denote the scaled, experimental, and quantum-chemically calculated values for X, respectively. In the present context, the scaling procedure can be used to provide very accurate predictions for rather high J, Ka rotational transitions (see, for example, Puzzarini et al. 2012). In Table 2, the measured rotational frequencies from Hirose (1974) are compared with our predictions; some predicted frequency values in the 80–250 GHz range are also collected (the complete list of prediction up to 500 GHz is provided in the supplementary material). From the comparison, we note that the calculated transitions are usually underestimated, with deviations ranging from 1 to 30 MHz and an averaged relative error of 0.02%. Therefore, we consider our predictions sufficiently accurate for laboratory or astronomical assignments. The predicted frequency values were obtained from the scaled rotational parameters using the SPCAT/SPFIT program (Pickett 1991).
Table 2. Comparison of Experiment and Theory for the Rotational Spectrum of Oxirane-d1
| Transition | Calculated | Experiment | |||||
|---|---|---|---|---|---|---|---|
| J' |  |
 |
J'' |  |
 |
||
| 1 | 1 | 0 | 1 | 0 | 1 | 10923.0 | 10925.00 |
| 2 | 2 | 0 | 2 | 1 | 1 | 16815.1 | 16822.39 |
| 3 | 2 | 1 | 3 | 1 | 2 | 21211.1 | 21212.88 |
| 2 | 1 | 1 | 2 | 0 | 2 | 21285.7 | 21284.05 |
| 4 | 3 | 1 | 4 | 2 | 2 | 25433.0 | 25443.48 |
| 3 | 3 | 0 | 3 | 2 | 1 | 27339.5 | 27356.03 |
| 5 | 3 | 2 | 5 | 2 | 3 | 31523.4 | 31522.22 |
| 2 | 2 | 1 | 2 | 1 | 2 | 32768.4 | 32774.05 |
| 6 | 4 | 2 | 6 | 3 | 3 | 33765.1 | 33776.67 |
| 4 | 2 | 2 | 4 | 1 | 3 | 33896.3 | 33890.10 |
| 5 | 4 | 1 | 5 | 3 | 2 | 35024.2 | 35048.15 |
| 1 | 1 | 1 | 0 | 0 | 0 | 37577.5 | 37580.17 |
| 3 | 1 | 2 | 3 | 0 | 3 | 38195.6 | 38190.67 |
| 3 | 3 | 1 | 3 | 2 | 2 | 40126.1 | 40137.71 |
| 4 | 4 | 0 | 4 | 3 | 1 | 41638.6 | 41664.47 |
| 7 | 5 | 2 | 7 | 4 | 3 | 42021.7 | 42052.25 |
| 8 | 5 | 3 | 8 | 4 | 4 | 42236.7 | 42232.50 |
| 7 | 4 | 3 | 7 | 3 | 4 | 42240.1 | 42246.24 |
| 2 | 0 | 2 | 1 | 1 | 1 | 58339.1 | 58336.18 |
| 4 | 2 | 3 | 4 | 1 | 4 | 59364.5 | 59363.97 |
| 3 | 0 | 3 | 2 | 1 | 2 | 87497.5 | |
| 3 | 1 | 3 | 2 | 0 | 2 | 89418.4 | |
| 3 | 2 | 2 | 2 | 1 | 1 | 112730.0 | |
| 4 | 0 | 4 | 3 | 1 | 3 | 114893.5 | |
| 4 | 1 | 4 | 3 | 0 | 3 | 115339.3 | |
| 4 | 1 | 3 | 3 | 2 | 2 | 127344.9 | |
| 3 | 3 | 1 | 2 | 2 | 0 | 136041.0 | |
| 4 | 2 | 3 | 3 | 1 | 2 | 136508.3 | |
| 3 | 3 | 0 | 2 | 2 | 1 | 141475.3 | |
| 5 | 0 | 5 | 4 | 1 | 4 | 141714.0 | |
| 5 | 1 | 5 | 4 | 0 | 4 | 141800.6 | |
| 5 | 1 | 4 | 4 | 2 | 3 | 157380.4 | |
| 5 | 2 | 4 | 4 | 1 | 3 | 160394.9 | |
| 6 | 0 | 6 | 5 | 1 | 5 | 168396.0 | |
| 6 | 1 | 5 | 5 | 2 | 4 | 185082.2 | |
| 6 | 2 | 5 | 5 | 1 | 4 | 185845.0 | |
| 7 | 0 | 7 | 6 | 1 | 6 | 195050.2 | |
| 7 | 1 | 7 | 6 | 0 | 6 | 195052.7 | |
| 7 | 1 | 6 | 6 | 2 | 5 | 211982.1 | |
| 7 | 2 | 6 | 6 | 1 | 5 | 212147.0 | |
| 8 | 0 | 8 | 7 | 1 | 7 | 221698.8 | |
| 8 | 1 | 8 | 7 | 0 | 7 | 221699.2 | |
| 7 | 3 | 5 | 6 | 2 | 4 | 231201.0 | |
| 5 | 5 | 0 | 4 | 4 | 1 | 235947.9 | |
| 9 | 0 | 9 | 8 | 1 | 8 | 248345.6 | |
| 9 | 1 | 9 | 8 | 0 | 8 | 248345.7 | |
Notes. Selected newly predicted rotational transition frequencies are also reported. All values in MHz.
Only a portion of this table is shown here to demonstrate its form and content. A machine-readable version of the full table is available.
Download table as: DataTypeset image
3.2. Vibrational Spectroscopy
Despite the significant experimental effort taken over the years, until 2012 only one band, namely, the υ3 fundamental, was analyzed at high resolution (Russell & Wesendrup 2003). The situation improved in the last couple of years thanks to the investigations by Lafferty et al. (2013; Flaud et al. 2012; Kwabia Tchana et al. 2013, 2014). The latter essentially confirmed the gas-phase wavenumbers reported by Nakanaga (1980), and provided accurate information on the υ1 and υ9 fundamentals, whose accurate assignment faced some difficulties due to overlapping transitions. However, a full accurate characterization of the IR spectrum of oxirane in terms of band positions and intensities, also including overtones and combination bands, is still lacking. In particular, there are no accurate data for the υ4, υ6, υ7, υ8, υ11, and υ14 fundamentals as well as for the υ1 intensity, which is overlapped with υ9 and υ13. As an example, we mention the υ4 band for which experimental values differing by about 40 cm−1 were reported in the gas phase (1109 cm−1 and 1148 cm−1 by Yoshimizu et al. 1975 and Bertie & Othen 1973, respectively). Today, the accuracy of computational approaches is such that they can be used to support and complement experimental studies, thus shedding light on controversial experimental data (Barone et al. 2013). Returning to the υ4 example, our best estimate of 1115.3 cm−1 tends to validate the result by Yoshimizu et al. (1975). Table 3 demonstrates the accuracy that can be reach by state-of-the-art computational models: a very good agreement between our best-estimated predictions (based on the CC(Best)/CC(VQZ) anharmonic force field) and experimental high-resolution data is evident. For line positions, the mean absolute error and maximum discrepancy are 5.5 cm−1 and 10.5 cm−1, respectively. We also note that essentially the same accuracy (within 1 cm−1) is obtained once the anharmonic corrections are evaluated at the CCSD(T)/cc-pVTZ level (CC(Best)/CC(VTZ) hybrid force field). This outcome is particularly important because the latter is the hybrid force field used for the mono-deuterated isotopologue of oxirane.
Table 3. Experimental and Computed Best Estimates for the Oxirane Fundamental Transitions
| Mode | Symm. | Assignment | Experiment | Theorya (This Work) | |||
|---|---|---|---|---|---|---|---|
| ν (cm−1) | I b (km mol−1) | ν (cm−1) | I (km mol−1) | λ (μm) | |||
| υ1 | A1 | CH2 sym stretch | 3018.35c | 3021.5 | 15.89 | 3.31 | |
| υ2 | A1 | CH2 scissor | 1497.83d | 0.92(0.06)e | 1490.0 | 1.19 | 6.71 |
| υ3 | A1 | ring breathing | 1270.37d,f | 13.16(0.45)g | 1266.2 | 13.57 | 7.90 |
| υ4 | A1 | CH2 wagging | 1115.3 | 0.22 | 8.97 | ||
| υ5 | A1 | Ring deformation | 876.7d | 64.76(3.6)h | 873.5 | 68.87 | 11.45 |
| υ6 | A2 | CH2 asym stretch | 3064.8 | 0.00 | 3.26 | ||
| υ7 | A2 | CH2 rocking | 1160.4 | 0.00 | 8.62 | ||
| υ8 | A2 | CH2 twisting | 1027.2 | 0.00 | 9.73 | ||
| υ9 | B1 | CH2 asym stretch | 3006.5c | 44.31(5.95)g | 3008.9 | 31.41 | 3.32 |
| υ10 | B1 | CH2 scissor | 1471.35d | 0.25(0.06)e | 1460.8 | 0.13 | 6.85 |
| υ11 | B1 | CH2 wagging | 1154.0 | 0.25 | 8.67 | ||
| υ12 | B1 | Ring deformation | 822.27d | 8.56(0.80)g | 815.9 | 8.32 | 12.26 |
| υ13 | B2 | CH2 asym stretch | 3066.0c | 36.24(4.69)g | 3076.0 | 34.19 | 3.25 |
| υ14 | B2 | CH2 twisting | 1145.4 | 4.13 | 8.73 | ||
| υ15 | B2 | CH2 rocking | 808.13d | 0.21(0.21)g | 810.2 | 0.25 | 12.34 |
Notes. aBest-estimated CC(Best)/CC(VQZ) vibrational wavenumbers and IR intensities. See text. bNumbers given in parenthesis are the experimental errors. cLafferty et al. (2013). dFlaud et al. (2012). eKwabia Tchana et al. (2013). fRussell and Wesendrup (2003). gNakanaga (1981). hKwabia Tchana et al. (2014). Total intensity in the spectral region 750–950 cm−1 corresponding to the υ5, υ12 and υ15 fundamental transitions.
Download table as: ASCIITypeset image
As far as intensities are concerned, it is evident that the theoretical values are only in fair agreement with experimental estimates, but it should be pointed out that most of the data come from low-resolution experiments (Nakanaga 1981) and are affected by rather large error bars. However, theoretical results agree within 10% at most with the high-resolution results available for υ2, υ5, and υ10. Furthermore, we note that the reliability of our computed IR intensities has been recently well demonstrated not only for fundamentals, but also for overtones and combination bands (Carnimeo et al. 2013; Pietropolli Charmet et al. 2013).
In view of supporting future experimental studies, predictions for the most intense transitions, including overtones and combination bands up to the two quanta, are reported in Table 4 for the oxirane and its mono-deuterated analogue. Based on the results presented in Table 3 and the literature on this topic (see, for example, Pietropolli Charmet et al. 2013), we can claim an accuracy of about 5 cm−1–10 cm−1 and 10 cm−1–20 cm−1 for fundamental and non-fundamental transitions, respectively. The latter error estimate for overtones and combination bands is also supported by the good agreement between the theoretical (2921.9 cm−1) and experimental (2930.8 cm−1) wavenumbers for the υ2+υ10 transition of oxirane.
Table 4. Best Estimates of the Vibrational Wavenumbers, Wavelengths, and IR Intensities for Oxirane and Oxirane-d1 in the 700–3100 cm−1 Region of Spectra
| Oxirane C2H4Oa | Oxirane -d1 C2H3DOb | ||||||
|---|---|---|---|---|---|---|---|
| Assignment | ν (cm−1) | λ (μm) | I c (km mol−1) | Assignment | ν (cm−1) | λ (μm) | I c (km mol−1) |
| υ13(CH2 a stretch) | 3076.0 | 3.25 | 34.19 | υ4+υ13 | 3084.7 | 3.24 | 0.18 |
| υ6(CH2 a stretch) | 3064.8 | 3.26 | 0.00 | υ4+υ14 | 3066.8 | 3.26 | 5.10 |
| υ1(CH2 s stretch) | 3021.5 | 3.31 | 15.89 | υ1(CH2 a stretch) | 3065.3 | 3.26 | 23.34 |
| υ9(CH2 a stretch) | 3008.9 | 3.32 | 31.41 | υ2(CH stretch) | 3029.6 | 3.30 | 9.89 |
| 2υ2 | 2952.7 | 3.39 | 0.10d | υ3(CH2 s stretch) | 3012.2 | 3.32 | 23.25 |
| υ2+υ10 | 2921.9 | 3.42 | 14.70e | υ5+υ9 | 2600.4 | 3.85 | 0.36 |
| 2υ10 | 2912.6 | 3.43 | 0.73 | υ6+υ11 | 2396.7 | 4.17 | 0.30 |
| υ10+υ11 | 2612.7 | 3.83 | 0.19 | υ5+υ13 | 2302.7 | 4.34 | 0.47 |
| υ2+υ4 | 2599.7 | 3.85 | 0.36 | 2υ8 | 2291.1 | 4.36 | 0.91 |
| υ10+υ5 | 2333.1 | 4.29 | 0.12 | υ5+υ14 | 2287.7 | 4.37 | 0.44 |
| 2υ11 | 2307.0 | 4.33 | 0.21 | υ7+υ11 | 2286.2 | 4.37 | 4.17 |
| υ7+υ14 | 2306.0 | 4.34 | 0.17 | υ8+υ9 | 2278.3 | 4.39 | 0.39 |
| υ2+υ12 | 2297.8 | 4.35 | 0.34 | 2υ9 | 2255.2 | 4.43 | 2.81 |
| υ11+υ4 | 2257.7 | 4.43 | 1.03 | υ4(CD stretch) | 2251.3 | 4.44 | 2.78 |
| υ11+υ8 | 2180.3 | 4.59 | 0.09 | υ6+υ12 | 2245.4 | 4.45 | 0.31 |
| υ14+υ8 | 2168.6 | 4.61 | 0.07 | υ8+υ10 | 2221.1 | 4.50 | 0.19 |
| υ3+υ5 | 2128.8 | 4.70 | 0.35 | υ6+υ13 | 2184.2 | 4.58 | 0.34 |
| υ3+υ12 | 2074.6 | 4.82 | 0.43 | υ6+υ14 | 2171.0 | 4.61 | 0.38 |
| υ11+υ5 | 2023.9 | 4.94 | 0.86 | 2υ10 | 2154.9 | 4.64 | 0.14 |
| υ14+υ5 | 2015.2 | 4.96 | 0.10 | υ7+υ12 | 2131.4 | 4.69 | 0.19 |
| υ11+υ12 | 1967.0 | 5.08 | 0.14 | υ10+υ11 | 2116.5 | 4.72 | 0.20 |
| υ14+υ15 | 1956.0 | 5.11 | 0.05 | υ7+υ13 | 2073.8 | 4.82 | 0.22 |
| υ4+υ15 | 1928.3 | 5.19 | 0.36 | υ7+υ14 | 2059.1 | 4.86 | 0.26 |
| υ8+υ15 | 1839.7 | 5.44 | 0.14 | υ9+υ12 | 2021.4 | 4.95 | 0.28 |
| 2υ5 | 1744.4 | 5.73 | 0.28 | υ9+υ14 | 1946.3 | 5.14 | 0.23 |
| 2υ12 | 1625.8 | 6.15 | 1.72 | 2υ12 | 1778.1 | 5.62 | 0.13 |
| υ2(CH2 scissor) | 1490.0 | 6.71 | 1.19 | 2υ13 | 1658.7 | 6.03 | 0.56 |
| υ10(CH2 scissor) | 1460.8 | 6.85 | 0.02 | υ13+υ14 | 1645.8 | 6.08 | 0.48 |
| υ3(ring breath) | 1266.2 | 7.90 | 13.57 | 2υ14 | 1628.3 | 6.14 | 0.73 |
| υ7(CH2 rock) | 1160.4 | 8.62 | 0.00 | υ13+υ15 | 1535.2 | 6.51 | 0.37 |
| υ11(CH2 wag) | 1154.0 | 8.67 | 0.25 | υ5(CH2 scissor) | 1475.4 | 6.78 | 0.46 |
| υ14(CH2 twist) | 1145.4 | 8.73 | 4.13 | 2υ15 | 1404.3 | 7.12 | 0.32 |
| υ4(CH2 wag) | 1115.3 | 8.97 | 0.22 | υ6(CHD scissor) | 1358.4 | 7.36 | 1.91 |
| υ8(CH2 twist) | 1027.2 | 9.73 | 0.00 | υ7(ring breath) | 1246.7 | 8.02 | 10.54 |
| υ5(ring deform) | 873.5 | 11.45 | 68.87 | υ8(CH2 rock) | 1146.4 | 8.72 | 2.00 |
| υ12(ring deform) | 815.9 | 12.26 | 8.32 | υ9(CH2 wag) | 1131.8 | 8.84 | 0.07 |
| υ15(CH2 rock) | 810.2 | 12.34 | 0.25 | υ10(CH2 twist) | 1079.1 | 9.27 | 1.14 |
| υ11(CHD wag) | 1039.6 | 9.62 | 2.18 | ||||
| υ12(ring deform) | 891.7 | 11.21 | 39.80 | ||||
| υ13(CHD twist) | 831.4 | 12.03 | 32.37 | ||||
| υ14(ring deform) | 816.7 | 12.24 | 11.28 | ||||
| υ15(CHD rock) | 701.2 | 14.26 | 2.76 | ||||
Notes. All IR transitions with intensities greater than 0.05 km mol−1 for oxirane and greater than 0.1 km mol−1 for oxirane-d1 are reported. aBest-estimated CC(Best)/CC(VQZ) vibrational wavenumbers and IR intensities. See text. bBest-estimated CC(Best)/CC(VTZ) vibrational wavenumbers and IR intensities. See text. cThe conversion factor to cm molecule−1 is cf = 6.023×1018 (i.e., values in km mol−1 should be divided by cf to obtain the corresponding values in cm molecule−1). dInvolved in the Fermi-II interaction with the υ1, total intensity ascribed to the υ1 can be possibly distributed over these two transitions. eInvolved in the Fermi interaction with the υ9.
Download table as: ASCIITypeset image
3.3. Astrophysical Implications
As pointed out by Bernstein & Lynch (2009), improved quantitative simulations of the UIR spectrum requires a more accurate set of Einstein spontaneous emission coefficients, A. The latter can be straightforwardly derived from the anharmonic intensities reported in Table 4. It is worth noting that our computations provide a twofold improvement: our IR intensities account for anharmonic contributions and, consequently, information on overtones and combinations bands can also be provided. Based on our computations, we simulated the entire UIR spectrum (800–6000 cm−1) and we compared it with the corresponding observations, taking as an example the ISO-SW spectrum of the planetary nebula NGC 7027 (Sloan et al. 2003). The portion corresponding to the 3–5.5 μm wavelength range is depicted in Figure 1, along with all identified line fluxes reported by Salas et al. (2001). We note that the intense features of oxirane, related to the CH2 stretching fundamental vibrations along with the υ2+υ10 overtone, match well the observed UIR features at 3.3 and 3.4 μm, for both the band position and their absolute intensities. However, it should be noted that the CH stretching vibrations are not characteristic of oxirane, and the broad features present in the IR spectra of circumstellar and interstellar nebulae suggest the presence of more complex mixed aromatic and aliphatic molecules (Kwok & Zhang 2011). On the other hand, the 11.2–11.3 μm UIR features match well (within 10 cm−1) the simulated band at 11.45 μm, which is related to the ring deformation, which can be considered more characteristic. Furthermore, we note that the simulated anharmonic spectra allow us to analyze also the features related to non-fundamental transitions, as those in the 4–5.5 μm range shown in Figure 1. It is noteworthy that several transitions of oxirane match the unassigned low intensity bands. As an example, the band pattern at about 4.35 μm due to the 2υ11, υ7+υ14, and υ2+υ12 transitions is highlighted.

Figure 1. Comparison in the 3–5.5 μm wavelength range between the simulated emission spectra of oxirane (red, lower trace) and the observed spectra (black, upper trace; from the uniform database of 2.4–45.4 μm ISO-SW spectrum of planetary nebula NGC 7027; Sloan et al. 2003) is shown. Identified line fluxes reported by Salas et al. (2001) are marked by gray lines. Theoretical spectra were convoluted with a Gaussian function with the half-width at half maximum of 1 cm−1.
Download figure:
Standard image High-resolution image{kind=link}
Moving to the possible identification of oxirane in Titan's atmosphere, we point out that the analysis of its IR spectrum is not a valuable route. The IR spectra recorded by Cassini (CIRS instrument; 10–1500 cm−1) show a strong absorption at 800–860 cm−1 due to the ethane υ9 band, which hampers the observation of the strongest oxirane transition (υ5), at 873 cm−1 (experimental value: 876.72 cm−1; Lafferty et al. 2012), in the CIRS working range. Furthermore, the modeling of the CIRS spectrum with a radiative transfer model not accounting for oxirane does not show any unassigned transitions in the residuals (i.e., the observed − calculated differences).
For the possible detectability of the oxirane molecule on Titan, one might rely on the assignment of its rotational spectrum. The Herschel Space Observatory with its three instrumentations, namely, SPIRE (Spectral and Photometric Imaging Receiver; 447–1550 GHz), HIFI (Heterodyne Instrument for the Far Infrared; 480–1250 GHz, and 1410–1910 GHz) and PACS (Photodetecting Array Camera and Spectrometer; 1363–5878 GHz), provides this opportunity. In particular, the HIFI instrument permits continuous-frequency high-resolution spectral surveys, and their analysis, assignment, and interpretation are only at the beginning. Even higher is the spectral resolution that can be exploited by the Atacama Large Millimeter Array, which is therefore expected to play a key role in the detection of complex species as well as rare isotopologues. It is in this context that we consider very useful our predictions of the rotational spectra of oxirane and oxirane-d1 in the submillimter-wave regime.
4. CONCLUDING REMARKS
State-of-the-art composite schemes have been used to compute highly accurate vibrational frequencies and spectroscopic constants for oxirane and its mono-deuterated isotopologue. The approaches followed are well tested and documented in the literature, and they are known to provide the required accuracy for guiding experimental investigations of rotational and IR spectra (see, for example, Puzzarini et al. 2010, 2012; Pietropolli Charmet et al. 2013; Barone et al. 2014). We are therefore confident that the present work is useful for shedding light on still open issues, like the assignment of unidentified IR bands or the observation of oxirane in Titan's atmosphere or the possible existence of the mono-deuterated isotopologue in the interstellar medium.
This work was supported by Italian MIUR (PRIN, FIRB) and by the University of Bologna (RFO funds). The high performance computer facilities of the DREAMS center (http://dreamshpc.sns.it) are acknowledged for providing computer resources. The support of COST CMTS-Action CM1002 "COnvergent Distributed Environment for Computational Spectroscopy (CODECS)" is also acknowledged. The research leading to these results has received funding from the European Union's Seventh Framework Programme (FP7/2007–2013) under grant agreement No. ERC-2012-AdG-320951-DREAMS.
The authors gratefully acknowledge Dr. A. Ali and Dr. C. A. Nixon (NASA Goddard Space Flight Center) for useful discussions on the assignment of Cassini CIRS spectrum.
Footnotes
- 4
CFour, a quantum chemical program package written by J. F. Stanton, J. Gauss, M. E. Harding, P. G. Szalay, with contributions from A. A. Auer, R. J. Bartlett, U. Benedikt, et al. and the integral packages MOLECULE (J. Almlöf, & P. R. Taylor), PROPS (P. R. Taylor), ABACUS (T. Helgaker, H. J. Aa. Jensen, P. Jørgensen, & J. Olsen), and ECP routines by A. V. Mitin & C. van Wüllen. For the current version, see http://www.cfour.de.