


ABSTRACT
Using a low-temperature 22-pole ion trap apparatus, detailed measurements for the title reaction have been performed between 10 K and 100 K in order to get some state specific information about this fundamental hydrogen abstraction process. The relative population of the two lowest H2 rotational states, j = 0 and 1, has been varied systematically. NH+ formation is nearly thermo-neutral; however, to date, the energetics are not known with the accuracy required for low-temperature astrochemistry. Additional complications arise from the fact that, so far, there is no reliable theoretical or experimental information on how the reactivity of the N+ ion depends on its fine-structure (FS) state 3Pja. Since in the present trapping experiment, thermalization of the initially hot FS population competes with hydrogen abstraction, the evaluation of the decay of N+ ions over long storage times and at various He and H2 gas densities provides information on these processes. First assuming strict adiabatic behavior, a set of state specific rate coefficients is derived from the measured thermal rate coefficients. In addition, by recording the disappearance of the N+ ions over several orders of magnitude, information on nonadiabatic transitions is extracted including FS-changing collisions.
Export citation and abstract BibTeX RIS
1. INTRODUCTION
As discussed recently by Dislaire et al. (2012) atomic nitrogen and nitrogen-containing molecules are important tracers for understanding astrophysical objects. Due to the abundance of hydrogen, the nitrogen hydrides, NH, NH2, and NH3, and their ions are of central importance in astrochemistry. For example, subsequent hydrogen abstraction reactions, starting with N+, finally lead to the formation of interstellar ammonia (Le Bourlot 1991). In order to obtain quantitative abundances in various environments, e.g., in dark clouds, the reaction
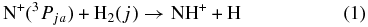
and its dependence on the rotational (j = 0, 1, ...) and the FS (ja = 0, 1, and 2) states plays a special role. In the 1980s and early 1990s, there were several experimental activities in which this reaction was studied at room temperature and below. Various techniques such as SIFDT (selected ion flow and drift tube; Adams & Smith 1985), CRESU (Cinétique de réactions en écoulement supersonique uniforme; Marquette et al. 1988), low-temperature penning ion trap (Barlow et al. 1986), and guided ion beams with scattering cells (Sunderlin & Armentrout 1994) provided detailed results. First applications of low-temperature radio frequency (RF) ion traps to the title reaction were mentioned in Gerlich (1989) and extended results have been given in Gerlich (1993). Probably the most sophisticated experiments have been based on crossing or merging a guided N+ ion beam with a supersonic hydrogen beam (Tosi et al. 1994); however, the ambitious goal of determining state specific cross sections has not been reached so far. There have also been various theoretical studies (see Gerlich 1989; Nyman & Wilhelmsson 1992; Wilhelmsson & Nyman 1992; Russell & Manolopoulos 1999, and references therein), and interesting aspects have been discussed; however, concerning reactions at low temperature, they raise more questions than they answer.
A basic problem is that one does not yet know whether the formation of NH+ + H is really endothermic or whether somewhere a barrier hinders the reaction. As reviewed by Gerlich (2008a), the analysis of measured temperature dependencies of reaction (1) with a statistical theory leads to an endothermicity of 17 meV if one presupposes that FS and rotational states are in thermal equilibrium and that, in promoting the reaction, their energies are as equally efficient as translational energy. If FS energy is not available at all, the assumed endothermicity has to be lowered to 11 meV to match the experimental results. High-level ab initio computations are not yet sufficiently accurate to predict this value with the required accuracy, i.e., within a few meV. This is rather unexpected since one has to calculate only the binding energy of NH+. Discussions of different high-level ab initio computations, calculated potentials, bond dissociation energies, and heat of formation for NH and NH+ can be found in Tarroni et al. (1997), Jursic (1998), and Amero & Vazquez (2005). Also related to this subject are the quantum chemical calculations of the adiabatic ionization energy of the NH2 radical (Willitsch et al. 2006), the accuracy of which has been estimated to be about 100 cm−1.
In order to understand reaction dynamics, one needs more than just the asymptotic energies. In the present case, several potential energy surfaces are involved. The situation is illustrated in Figure 1 with a simplified electronic correlation diagram (Mahan & Ruska 1976; Russell & Manolopoulos 1999). Inspection reveals that, at kinetic energies below 1 eV, only the NH+(X2Π) product can be formed, if the two reactants N+ and H2 are in their ground state. For Cs symmetry the reaction can proceed adiabatically via the more than 6 eV deep double-well structure, as indicated by the dashed lines avoiding the crossings. An analytical fit of this lowest adiabatic 3A'' potential energy surface has been published by Wilhelmsson et al. (1992). This surface has been used for low-energy quasi-classical trajectory calculations (Nyman & Wilhelmsson 1992; Wilhelmsson & Nyman 1992) as well as for time-dependent wave packet studies (Russell & Manolopoulos 1999). Problematic for the comparison of calculated reaction probabilities with low-temperature experimental data is that, on this surface, the reaction endothermicity is certainly too large with 33 meV.

Figure 1. Schematic view of the electronic state correlation diagram for the NH2+ system (not to scale; for details see Mahan & Ruska 1976; González et al. 1986; Russell & Manolopoulos 1999). The horizontal dashed line indicates that reaction (1) is nearly thermoneutral. Typical experimental and theoretical values for the endothermicity (or barrier) range from 11 meV (Gerlich 2008a) to 33 meV (Wilhelmsson et al. 1992). The formation of a strongly bound NH2+ complex is possible via avoided intersections (Cs symmetry; dashed lines). Important for low-temperature collisions is the coupling between the nine near-degenerate spin–orbit potential-energy surfaces during the approach of the reactants. This can lead both to FS changes and reaction. The situation is illustrated in the lower left corner on a magnified scale in adiabatic approximation but also indicating schematically nonadiabatic transitions. The potential curves have been estimated in analogy to the N+–He collision complex (Soldan & Hutson 2002).
Download figure:
Standard image High-resolution imageAnother shortcoming of the calculations mentioned above is that the role of FS splitting is included using only rather crude approximations. As can be seen from the electronic correlation diagram in Figure 1, only one of the three triplet surfaces that correlates with the entrance channel, allows direct access to the deep well, while the other two are repulsive. In statistical theories, this is accounted for by using suitable electronic degeneracy factors concerning the N+ + H2 system this is discussed in Gerlich (1989). A closer look at the initial splitting region (small circle in Figure 1) reveals that the open-shell structure of N+(3Pja) results in at least nine spin–orbit coupled potential-energy surfaces. As plotted schematically in Figure 1 (large circle), they converge at large distances toward the three states 3P0, 3P1, and 3P2. The question remains of how one can reach the product channel on these nine surfaces. Is there access to the deep well without any activation barrier? The assumption that only the three lowest spin–orbit surfaces lead to products, while reaction on the other six is not possible at low energies is very restrictive. This model, which is based on strict adiabatic behavior, was applied by Wilhelmsson & Nyman (1992) and Nyman & Wilhelmsson (1992). It has also been used by Russell & Manolopoulos (1999). Therefore, we also start the evaluation of our data on this basis. However, strict adiabatic behavior not only forbids reactions of ions in the 3P2 state but also inhibits FS-changing collisions. As a consequence, N+ ions in the highest fine structure would be completely insensitive to collisions with H2. Since in our experiment, however, all N+ ions are hydrogenated sooner or later, a more sophisticated kinetic model is required to account for the competition between reaction and thermalization of all three FS states.
Disregarding the details of any multi-surface model, a simpler question is how the different forms of energy, stored in the excited states of the reactants, can help to promote the reaction in the endothermic direction. Comparison of the thermal motion of the reactants (here up to 100 K, corresponding to 3/2 kB T = 12.9 meV) with the electronic energy of N+ (3P1 6.1 meV, 3P2 16.2 meV) and rotational energy of H2 (14.4 meV for j = 1) reveals that all these values are comparable to the endothermicity or barrier (11 or 17 meV, see above).
The equivalence of rotational and translational energy was first shown in the pioneering work from Marquette et al. (1988). In their low-temperature flow experiment, reaction (1) was studied using para hydrogen (p-H2) as well as normal hydrogen (n-H2). Note that p-H2 has a total nuclear spin I = 0 and only even rotational states are allowed while ortho hydrogen has I = 1 and odd rotational states. Normal hydrogen is the 300 K statistical mixture consisting of 1/4 p-H2 and 3/4 o-H2. In what follows, other mixtures are characterized with the abbreviation f, indicating the fraction of o-H2. The results from Marquette et al. (1988) were corroborated in a low-temperature trapping experiment (Gerlich 1993) and were extended using mixtures with f = 0.13, 0.03, and <0.01. More systematic studies of the f dependence are reported in this work.
In all experimental papers prior to 1994 it was postulated that the energy of the excited FS states is equivalent to translational and rotational energy. In addition it has been assumed that, in flow (Marquette et al. 1988) or trapping experiments (Gerlich 1993), thermal populations of the 3Pja states are reached rather quickly. There has never been any proof of this. In a guided ion beam experiment (Sunderlin & Armentrout 1994), FS energy was simply accounted for using the thermal mean value (9.5 meV at 300 K). As already mentioned, an analysis of all these experimental results, based on a detailed statistical model (Gerlich 1989), came to the conclusion that 17 meV is needed to promote the reaction. First doubts concerning the efficiency of the FS energy were formulated in Tosi et al. (1994), especially in the context of Figure 4 of that publication.
This contribution reports new experimental results measured with a variable temperature RF ion trap. After a brief description of the instrument and typical measuring and calibration procedures, different sets of data are presented including the dependence of rate coefficients on the temperature and on the ortho fraction f and the time dependence of converting primary ions into products. The data are evaluated using first the adiabatic model resulting in state specific rate coefficients kj,ja(T) for j = 0, 1 and ja = 0–2. In Section 4, additional information on the reactivity of ja = 2 and the FS relaxation rate coefficient is presented. Some remarks concerning planned and possible extension of this work will conclude this paper.
2. EXPERIMENTAL
The instrument used in this study is the Chemnitz AB 22 pole trapping apparatus (Gerlich et al. 2011) which has been operated since 2009 at Charles University in Prague. It has been used recently in combination with an effusive beam of H atoms (Plasil et al. 2011; Gerlich et al. 2012). In the present study, the neutral target gas is leaked directly into the trap. The basics of storing ions in RF fields have been described thoroughly in Gerlich (1992, 1995). A summary of typical applications in low-temperature ion chemistry has been given in Gerlich (2008a, 2008b).
The central part of the instrument is shown schematically in Figure 2. The trap (22 rods with 1 mm diameter) is surrounded by a copper box which is mounted onto the cold head of a closed-cycle helium refrigerator. Stationary temperatures between 10 K and 100 K are set by simultaneously cooling and heating. Alternatively, temperature-dependent measurements are performed during the cooling down or warming up phases of the cold head. Hydrogen gas can be introduced into the trap via two leak valves, allowing us to produce any mixture from almost pure p-H2 (f < 0.01) to n-H2 (f = 0.75). A few collisions of the neutral gas with the walls are sufficient to get it into thermal equilibrium with the trap temperature, with the exception of the ortho/para ratio. The gas density inside the trap is determined using a spinning rotor gauge or a calibrated ionization gauge. The background pressure of the main chamber is lower than 10−7 Pa. With the exception of HD, most gas impurities are frozen out below 100 K; nonetheless, the small concentrations left can lead to errors as discussed below.

Figure 2. Schematic view of the 22 pole ion trap instrument used for studying reaction (1). The copper box surrounding the trap can be cooled down to 10 K. N+ ions are produced from N2 gas in a storage ion source (not shown), using electrons with a kinetic energy of 60 eV. After mass selection, they are transferred to the trap via an electrostatic quadrupole bender. In the radial direction the ions are confined by the RF field (Ω/2π = 19 MHz, V0 = 20 V). The potential inside the trap can be corrected locally with five ring electrodes. The entrance and exit electrode are used to open and close the trap with electrostatic barriers of some tens of meV. To the right, ions move through the quadrupole mass spectrometer toward the detector.
Download figure:
Standard image High-resolution imageThe primary N+ ions are produced via dissociative ionization of N2 in a storage ion source using energetic electrons (60 eV). Under such conditions it is safe to assume that the three fine-structure states 3P0, 3P1, and 3P2 are populated according to their statistical weights, i.e., with 1, 3, and 5, respectively. Attempts to thermalize this population prior to reaction have been described in Gerlich (1993) and Tosi et al. (1994); however, no changes in the reactivity have been observed. After passing a mass filter and an electrostatic quadrupole bender, the primary ions are transferred into the trap. During the filling period, the electrostatic barrier at the entrance electrode is slightly negative relative to the potential of the trap. After various storage times, the trap exit is opened and the ions move through the quadrupole mass spectrometer and are converted into a fast negative pulse using an MCP detector followed by a discriminator. The standard measuring procedure is based on filling the trap at a fixed frequency with a well-defined number of primary ions (typically a few thousand) and analyzing the content after different storage times.
A typical set of raw data, recorded at a 22 pole temperature of 52 K and with buffer and reactant gas in the trap ([He] = 5.2 × 1011 cm−3, [n-H2] = 1.6 × 1011 cm−3), is shown in Figure 3. The injected N+ ions react with hydrogen and form NH+. In subsequent collisions, these products react to form NH2+ and finally NH3+. As discussed by Gerlich (1993), NH4+ is also formed at length, however, very slowly, most probably via tunneling. In the first 10 ms, the sum of all detected ions (Σ) is increasing. This is due to phase-space compression of the injected ion cloud via collisions with the cold buffer gas leading to an increase of the detection efficiency (mainly acceptance and transmission of the quadrupole). Usually such time dependences are fitted with the solutions of a suitable rate equation system. In the present study, most information is derived simply from the decay of the primary ions. Quick information on the decay time constant is obtained by recording the number of N+ ions at two or three suitable storage times. In order to get deeper insight into the kinetics, the disappearance of N+ is followed in small time steps over several orders of magnitude (see below).

Figure 3. Sequential hydrogenation of trapped N+ ions leading to NHi+ (i = 1–3) as a function of the storage time t. The measurements were performed at 52 K with helium buffer gas [He] = 5.2 × 1011 cm−3 and at a hydrogen number density of [n-H2] = 1.6 × 1011 cm−3. The sum of all ions (Σ) increases at the beginning (phase-space compression) and decays than very slowly. The 3.6% loss is most probably due to reactions with impurities. Evaluation of the decay of N+ leads to a mean rate coefficient of 2.67 × 1010 cm3 s−1.
Download figure:
Standard image High-resolution image3. RESULTS AND FIRST EVALUATION
The upper part of Figure 4 shows a collection of rate coefficients, measured for the title reaction between 100 K and 10 K for different o-H2 fractions f, ranging from 0.75 to 0.005. As explained in Section 2, most data have been taken during the cooling down phase of the cold head. In addition, some tests at selected temperatures have been made. Several sets of data, taken on different days, have confirmed the reproducibility.

Figure 4. Arrhenius plot of experimental rate coefficients k for reaction (1), measured at different ortho-fractions f (upper panel). The data can be reproduced quite well with a thermally weighted superposition of state specific rate coefficients. The used function is given in Equation (3), the parameters in Table 1. The thin dashed lines are the analytical results for f = 0 (pure p-H2) and f = 1 (pure o-H2). In the lower panel, our data are compared to previous results. The triangles are CRESU results (Marquette et al. 1988), while the dots are ion trap results reported in (Gerlich 1993). The data at room temperature and above have been measured with an SIFDT instrument (Adams & Smith 1985). The dashed lines which go through the triangles indicate the functions used recently by Dislaire et al. (2012).
Download figure:
Standard image High-resolution imageOne approach to evaluating such a manifold of experimental results is to use simple Arrhenius-type functions,
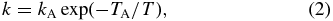
where TA = EA/k is the activation temperature, EA is the activation energy, and k is the Boltzmann constant. In order to limit the number of free parameters, the restrictions imposed by the adiabatic model mentioned in Section 1 have been implemented. This means only the lowest three FS states can lead to products while the other six are so repulsive that there is no low-energy reaction path. In addition, it is postulated that the population of the FS states relaxes efficiently to the temperature of the trap. This assumption has also been made in the evaluation of all previous experimental data (Marquette et al. 1988; Gerlich 1993), although there has never been a direct proof. Under these boundary conditions, the measured rate coefficient k can be fitted using the ansatz

In this equation, f is the selected ortho fraction, and the coefficients ξja account for the thermal population of the two lowest FS states ja. For the state specific rate coefficients, kj,ja(T) (with j = 0, 1 and ja = 0 and 1), individual Arrhenius functions have been chosen. Contributions from j = 2 (3% in pure p-H2 at 100 K) have been neglected. Inspection of the upper part of Figure 4 reveals that the lines follow the data points rather precisely, i.e., the restricted model fits all experimental data quite well. The parameters obtained are presented in Table 1. More aspects of this analysis will be discussed below.
Table 1. State Specific Rate Coefficients for the Title Reaction, kj,ja (units 10−10 cm3 s−1 and K), Derived from the k(T)
| j, ja | kA | TA |
|---|---|---|
| k0,0 | 12 | 230 |
| k1,0 | 1.9 | 18 |
| k0,1 | 14 | 230 |
| k1,1 | 12 | 40 |
Notes. Results measured for various ortho fractions f (see Figures 4–6) and using Equation (2). k0,2 and k1,2 are set to zero (adiabatic approximation) and it is assumed that the ions are thermalized. For more information see the text.
Download table as: ASCIITypeset image
In the lower part of Figure 4, our data are compared to a variety of previously published measurements. For clarity and orientation, only our new analytical fits for f = 0.75 and 0.05 are included as solid lines. In addition, Table 2 presents numerical values from other experiments and from a statistical theory. It can be seen that the previous ion trap results (Gerlich 1993) are slightly lower for n-H2 (f = 0.75); however, they overlap within the combined uncertainties of absolute rate coefficients (typically 20%). The agreement of the steep decay of k between 100 K and 40 K for almost pure p-H2, following the dotted line for j = 0 in the upper panel is gratifying. At temperatures below 25 K, the plots for the various mixtures run more or less parallel, indicating that k is mainly determined by the first half of Equation (3), i.e., the contributions from hydrogen in j = 1.
Table 2. Previous Measured and Calculated Rate Coefficients for the Title Reaction (units 10−10 cm3 s−1 and K)
| kA | TA | n | Remarks | Reference |
|---|---|---|---|---|
| 8.35 | 168.5 | 0 | p-H2 | Marquette et al. (1988) |
| 4.16 | 41.9 | 0 | n-H2 | |
| 15.3 | 177.5 | 0 | p-H2 | Gerlich (1989) |
| 4.06 | 42.5 | 0 | n-H2 (27–45 K) | |
| 14.0 | 230 | 0 | p-H2 | Gerlich (1993) |
| 1.1 | 26 | 0 | n-H2 (10–40 K) | |
| 4.2 | 44.5 | −0.17 | o-H2 | Dislaire et al. (2012) |
| 9.0 | 220 | 0 | p-H2 | |
| 1.5 | 180 | −2.1 | o-H2 | This work |
| 1.75 | 15 | 0 |
Note. For comparison, the results from this work (last three lines) have also been parameterized using Equations (2) and (5).
Download table as: ASCIITypeset image
As explained in the context of Figure 2, the f values have been set absolutely by mixing n-H2 and p-H2. This method leads to very reliable values for f > 0.2. An analysis of possible errors indicates, that mixtures with less o-H2 (f < 0.2) have an relative uncertainty of up to 10%. The ortho fractions given for "pure p-H2" (f < 0.01) have been determined by fitting the experimental data using f as a free parameter. Note, however, that it is not yet clear whether the loss of N+ in the low-temperature region is really just due to the reaction with the small o-H2 admixtures or whether other processes, e.g., tunneling through a barrier, gas impurities such as HD or RF heating, are the reason for this.
In addition to the systematic variation of the temperature, the f dependence of the rate coefficients has also been recorded by increasing the ortho fraction in small steps from near zero to 0.75. The results, obtained at five different temperatures, are plotted in Figure 5. Comparison with the dashed lines reveals that all data follow a linear increase in good approximation. This is in accordance with Equation (3) predicting such an f dependence; however, it is only valid within the simple model used. Possible deviations could be expected due to more complex kinetics occurring in the trap, especially due to differences in FS relaxation for collisions of N+ with H2(j = 0) or H2(j = 1). In this context, it is an interesting question whether the exothermic transfer of rotational energy into FS energy,
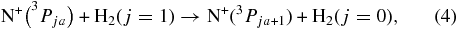
can affect the FS population at very low temperatures. Most probably, this process is forbidden by nuclear spin restriction, i.e., the required ortho–para conversion is very unlikely.

Figure 5. Rate coefficients for reaction (1) measured as a function of the ortho fraction for the indicated temperatures. As predicted from Equation (3) the data show a linear increase with increasing f. Nonetheless systematic deviations cannot be ruled out, especially at very low temperatures. It should be mentioned that the 11 K results extend over two orders of magnitude.
Download figure:
Standard image High-resolution image4. DISCUSSIONS
As long as there are no directly measured state specific cross sections, assumptions have to be made about the role of the different energy forms in driving the reaction. In the evaluation above, the reactivity of ions in the highest excited FS state has been set to zero, based on the adiabatic model, leading to very good fits of the data. This agreement, however, cannot be taken as a proof of the validity of this model, since other analytical functions, also used in the analysis of chemical systems, are of fitting capable them.
Deviations from the simple Arrhenius form (Equation (3)) are well known. They can be traced back to deviations of the threshold onset of an endothermic cross section from the functional form ∼(Et − Eo)1/2/Eo (Et is the translational energy; Eo is the threshold energy). In astrochemical data systems (see, for example, Equation (1) in Wakelam et al. 2012) it is common to account for this using a pre-exponential temperature-dependent factor with a free parameter n,
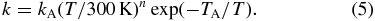
This function, which is called the Arrhenius–Kooij formula, has been used recently by Dislaire et al. (2012) for reevaluating the N+ + H2 data of Marquette et al. (1988). Ignoring the role of FS energy, they obtained a new set of parameters for o-H2 (see Table 2, Figures 4 and 6). For p-H2 they used the unaltered results reported by Marquette et al. (1988).

Figure 6. Two different fits of k(T) measured with n-H2. In the upper panel Equation (3) is used with the parameters given in Table 1. The four thin lines show the individual contributions kj,ja, weighted with the thermal population of the FS states (ξja) and the ortho fraction f. In the lower panel, the functions given in Equations (2) and (5) have been used directly. Three functions had to be used to get also a good fit. They are plotted as three thin lines together with the parameters used. In addition, this plot also shows the results from phase-space theory (short dashed line (Gerlich 1989) and the function used by Dislaire et al. (2012).
Download figure:
Standard image High-resolution imageIn Figure 6, the results of two different fitting procedures can be compared with each other. In the upper part, the f = 0.75 results from Figure 4 are reproduced together with the fit based on Equation (3). The four thin lines show the individual contributions kj,ja weighted with the thermal population of the FS states (ξja) and the ortho fraction f. Inspection of these contributions reveals that in this model the curvature of the measured data is mainly caused by the change of the thermal population of the FS states. Between 50 K and 100 K the state specific rate coefficient k1,1, i.e., reaction of N+(3P1) with H2(j = 1), prevails while the contribution from FS ground-state ions dominates at low temperatures. It is obvious that the results obtained for f = 0.75 are not very sensitive to k0,0 and k0,1. For these state specific rate coefficients more information has been derived from measurements with p-H2 as can be seen in Figure 4. In the lower panel, the three indicated functions (thin lines, based on Equations (2) and (5)) have been used. With the parameters given in the lower part of Table 2, very good agreement with the data points has also been reached. In this case, the curvature of the data is reproduced by the pre-exponential term. With the exception of the activation temperatures TA, a scientific interpretation of these results is not obvious. Perhaps this can be taken as a hint that the 3P1 state really plays a significant role at temperatures above 40 K.
The two examples shown in Figure 6 and discussed above illustrate that the analysis of the manifold of measured data is somehow arbitrary and that more experimental information is needed. Another problem, already mentioned in Section 1, is that our evaluation of the data is internally inconsistent since the 3P2 state is excluded postulating adiabatic behavior on one side but it assumes efficient FS relaxation on the other side. In order to shed some more light on this conflict, additional experimental information is used, namely, the temporal changes of the ion composition in the trap as illustrated and discussed in Figure 3 and shown in Figure 7.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. Decay of N+ ions measured at the indicated temperatures and for n-H2 (left, f = 0.75) and for p-H2 with minor o-H2 impurities (right, f < 0.01). Note the different scales. All relevant experimental parameters are collected in Table 3. The experimental data (circles) can be fitted well with a first-order exponential decay resulting in the mean rate coefficients 〈k〉 (also given in Table 3). In reality, the kinetics are more complicated due to the competition of FS-changing collisions and reactions with H2. The three thin lines, marked with 3Pja (ja = 0, 1, 2) show a special solution of the changes of the relative number of N+ ions in specific FS states. In all cases, the high-temperature ratio 5:3:1 has been assumed for the initial population. The state specific rate coefficients for reaction and relaxation are given in Tables 1 and 4. Note that this result is not unique.
Download figure:
Standard image High-resolution image{kind=link}
Since, due to the ionization process, the initially injected primary N+ ions are in the 3P2 state with a probability of 5/9 (3/9 in 3P1, 1/9 in 3P0) thermalization of the trapped ion ensemble and hydrogen abstraction reactions occur in competition with each other. In order to model the kinetics, one needs the relevant rate coefficients. A first attempt, based on state specific reaction rate coefficients calculated with phase-space theory (Gerlich 1989) and assuming relaxation rate coefficients krja→ja-1 = 10−n cm3 s−1 with n = 11, 10, and also 9, was reported in Tosi et al. (1994). While all measured decay curves were mono-exponential (after thermalizing the kinetic energy; see also Figure 4 of Gerlich 1993) the simulation always predicted an initial fast decay of primary ions followed by a slower one (see Figure 4 of Tosi et al. 1994). This curved behavior is obviously due to the high reactivity of the excited N+ ions as predicted by the phase-space theory. In Tosi et al. (1994), a trivial solution has been proposed: fine structure energy is just not available, i.e., all ions react with the same rate coefficient. Looking at the potential energy surface and Figure 1, there is no obvious reason for such an extreme behavior.
For testing various assumptions, additional measurements have been performed under different experimental conditions. The number density of H2 has been varied over more than two orders of magnitude. In addition, He buffer gas has been added with a number density of some 1012 cm−3 so far without any significant changes. A selection of new results is plotted in Figure 7. The upper part shows the decay of the relative number of N+ ions at about 100 K, the lower part of figure shows decay at 11 K. In the measurements on the left, n-H2 was been used; on the right p-H2 was used. All relevant experimental parameters are collected in Table 3. As can be seen from the thin lines, all four data sets can be fitted almost perfectly with a simple exponential decay function,

From the measured decay times τ and the hydrogen number densities [H2], mean rate coefficients have been calculated

The resulting parameters 〈k〉, which are an average over the time dependent FS population, are included in Table 3. In order to understand these observations in more detail, several additional facts need to be mentioned. (1) The decay rate of all N+ ions is at all times the same. The slight deviations during the thermalization of the translational energy after ion injection can be ignored at the timescales used in Figure 7. (2) The monotonous decay can be followed until storage times where only 10−4 of the injected ions are left over. (3) Simulations with rate coefficients from phase-space theory always lead to a faster decay at the beginning (concave curvature). (4) Setting the rate coefficient for the 3P2 state to zero (strict adiabatic model) but allowing for relaxation always leads to a convex curvature.
Table 3. Experimental Parameters Used in the Measurements Shown in Figure 7
| a | b | c | d | |
|---|---|---|---|---|
| T (K) | 110 | 98 | 11 | 11 |
| [H2] (cm−3) | 1.8E12 | 2.6E11 | 2.0E12 | 2.8E13 |
| f | 0.75 | 0.0055 | 0.75 | 0.0093 |
| [He] (cm−3) | 2.4E12 | 2.3E12 | 1.2E12 | 2.4E12 |
| 〈k〉 | 3.20E−10 | 1.00E−10 | 2.20E−11 | 2.20E−13 |
Download table as: ASCIITypeset image
Guided by these facts we have developed a kinetic model that includes reaction of all three states of N+ and also FS-changing collisions. In order to restrict the number of free parameters of the simulation, we started with the state specific rate coefficients derived form our experiment, i.e., with the values given in Table 1. Then the rate coefficients k0,2 and k1,2, which have been set to zero in the adiabatic model, have been increased slowly. Motivated by the interaction shown schematically in Figure 1 only FS transitions from 2 to 1 and 1 to 0 have been accounted for with the relaxation rate coefficients kr2→1 and kr1→0. The reverse rate coefficients, kr1→2 and kr0→1, have been included in the calculation, making use of micro-reversibility. Direct transitions between 2 and 0 are assumed to be inefficient.
It has been rather easy to find parameters describing each individual data set. It also became evident that there are many specific solutions although there is a strong correlation between the competing processes imposed by the mono-exponential decay of the experimental data. The relaxation and reaction rate coefficients strongly depend on each other. To our surprise we finally found a very simple solution, taking the four unchanged rate coefficients given in Table 1 and also using simple Arrhenius-type rate coefficients for the highest FS state and for relaxation.
The resulting parameters fitting all our data sets are given in Table 4. The solutions for the temporal changes of the number of N+ ions in specific 3Pja states and their sum are plotted in Figure 7. In all cases, the sum (solid line) follows nicely the mono-exponential decay of the measured data. A detailed inspection of (a) and (b) reveals that FS thermalization is achieved only after 30 ms since the relaxation rate coefficient is rather slow (4 × 10−11 cm3 s−1). In order to get the mono-exponential decay during this time the concave function describing the decay of ions in the 3P2 state is compensated by the two convex functions. At 11 K relaxation is even slower (2 × 10−12 cm3 s−1) and with n-H2 (panel (c)) reaction is faster than relaxation. In order to get the mono-exponential time dependence, the three rate coefficients are similar. Impressive is the result (d), where the N+ can only react with the traces of H2 (j = 1). Since in this case the H2 number density is more than 10 times higher than in (c), thermalization of the FS population is achieved in 200 ms. For further conclusions, more measurements and a detailed mathematical analysis of the coupled differential equations are needed.
Table 4. State Specific Rate Coefficients for Reaction, kj,ja, and Relaxation, krja→ja', Derived from the Fits Shown in Figure 7 (units 10−10 cm3 s−1 and K)
| kA | TA | |
|---|---|---|
| k0,2 | 0.51 | 53.4 |
| k1,2 | 1.44 | 18.4 |
| kr2→1 | ||
| 0.58 | 37.0 | |
| kr1→0 |
Note. Rate coefficients needed in addition to those in Table 1 for fitting the exponential decay shown in Figures 7(a)–(d).
Download table as: ASCIITypeset image
In summary, the results of our model allow several conclusions. (1) FS-changing collisions are rather slow, especially at low temperatures indicating nearly adiabatic behavior. (2) In contradiction to the strict adiabatic model, the highest FS state contributes to the formation of NH+ products, but much slower than predicted from statistical calculations. (3) Our experimental observations are qualitatively in accordance with the nonadiabatic couplings indicated in the oversimplified Figure 1. For a more quantitative understanding, one needs a detailed adiabatic or nonadiabatic formulation of the multi-surface problem. The first approach may be similar to the N+–He system (Soldan & Hutson 2002); however, in addition to the spin–orbit coupling, one must include the correct long-range attraction (charge-induced dipole and charge–quadrupole) and the couplings induced by the anisotropy, and one has to account for the effects caused by the rotation of H2. At very low energies hyperfine-interaction may also finally play a role. All this leads to a complex switch yard of crossings and needs help from theory to be sorted out!
5. CONCLUSIONS AND OUTLOOK
Based on an extensive set of new experimental rate coefficients k(T; f) as well as on a careful analysis of the decay curves of trapped N+ ions, for the first time state specific rate coefficients for the interaction of N+(3Pja) with H2(j) have been extracted. There are still uncertainties concerning the role of the FS energy. Nonetheless it is rather clear that excitation of N+ to the 3P2 state reduces its reactivity, but not completely to zero as predicted from strict adiabatic assumptions (Wilhelmsson & Nyman 1992; Russell & Manolopoulos 1999).
As long as astrochemical models ignore the FS states of the N+ ions, it is recommended that the rate coefficients for j = 0 and 1, presented in the lower part of Figure 6 and in Table 2, be used. However, it must be noted that the two N+ lines (3P2 → 3P1) at 121.9 μm and (3P1 → 3P0) at 205.2 μm play an important role in certain astrophysical environments, e.g., in photo-dissociation regions, where matter is heated via penetrating far-ultraviolet photons and cooled via forbidden atomic fine-structure transitions. A detailed discussion of such cooling lines observed in the Orion Bar can be found in a recent publication by Bernard-Salas et al. (2012). It is obvious that one needs detailed rate coefficients for inelastic and reactive collisions with electrons, atoms, and molecules in order to model such observations.
The presented experimental results give some first information on the state specific rate coefficients kj,ja(T) for all combinations of ja = 0–2 and j = 0–1. In order to check the results presented additional experiments must be performed. For example, using He number densities of several 1015 cm−3 and relaxation times of seconds or longer before hydrogen is leaked into the trap may finally lead to relaxation of the FS population prior to the reaction. An ultimate experiment would be the in situ state selected ionization of N-atoms via autoionizing resonances. Another striking idea is to maintain a stationary FS population of the trapped N+ ions using an intense microwave wave field at the wavelengths mentioned above.
As established in Gerlich et al. (2011) the apparatus used in this work can also be operated with a neutral target beam. To fully understand the NH2+ collision system we plan to study the reverse reaction
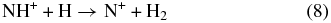
as a function of the temperature of the ion and the atomic beam source, similar to CH+ + H (Plasil et al. 2011). This beam-trap arrangement can also be used with a high-temperature accommodator for producing rotationally and vibrationally excited H2. Finally, it must be mentioned that the various deuterated variants of reactions (1) and (8) including the endothermic D–H exchange in ND+ + H will provide deep insight into the role of zeropoint energies, barriers, and tunneling at low energies.
Since 2010, the AB 22PT instrument has been operated at the Faculty of Mathematics and Physics of Charles University in Prague. We thank the Technical University of Chemnitz and the DFG for lending us this instrument. This work is a part of research grant OC10046 financed by the Ministry of Education of the Czech Republic and was partly supported by GACR (P209/12/0233, 205/09/1183), by GAUK 388811, GAUK 406011, and by COST Action CM0805 (The Chemical Cosmos).