

Abstract
Despite several studies on film-forming systems with the advantages of both the film and the hydrogel, there are still no effective systems for fast film formation with a high level of control over permeability. In this study, a film-forming system for the delivery of nanomedicine, termed a film-forming nanogel (FFN), was produced and investigated for the first time to meet this need. The objective of this research was to study a new generation of film-forming hydrogels (FFHs) loaded with curcumin nanoparticles (CUR-GNPs) for transdermal applications. FFHs were prepared by employing zein and HPMC 4000 as film-forming polymers. Meanwhile, CUR-GNPs were obtained by sonoprecipitation. The film-forming time, particle characteristics and FFN drug release profile were assessed. The optimized FFH had a smooth surface and a fast drying time of 6 min and 4.5 min in vitro and ex vivo, respectively. Additionally, high, sustained drug permeation from the FFN was observed after 24 h. The FFH containing CUR-GNPs showed potential for application in transdermal drug delivery with a fast film-forming time, uniform particle dispersion and high, sustained drug permeation.
Export citation and abstract BibTeX RIS
1. Introduction
Although oral administration is the most preferable route for drug delivery due to its convenience and patient compliance, there are problems related to pH variations in the GI tract and the hepatic first-pass metabolism, which result in low bioavailability at the target treatment site [1, 2]. On the other hand, injection, another common administration route, also has several disadvantages, including high cost, labour-intensive production, patient distress, local reactions, and risk associated with drugs with the propensity to form precipitates [3, 4]. Transdermal administration has attracted attention as a favourable route for drug delivery due to its convenience, painless self-administration, and avoidance of the hepatic first-pass metabolism and GI tract, among other features [5]. Moreover, skin has a high surface area of approximately 2 m2 and can feasibly be used for the application of transdermal treatments [6]. These outstanding properties of transdermal drug delivery render it a first-line option as it overcomes the majority of the shortcomings of oral and parenteral administration methods. However, the delivery of drugs through the skin has challenges associated with the skin barrier, which is composed of two layers: the epidermis and the dermis. Importantly, the key negative impact originates from the stratum corneum, the outermost layer of the epidermis, which modulates the absorption of compounds into deeper skin layers and hence further affects the penetration and permeation of drugs through the skin [7, 8]. Moreover, the transdermal delivery of therapeutic agents may be unintentionally decreased due to sweat or an external aqueous environment [9], thus resulting in treatment failure. This study demonstrates a solution to overcome these hurdles.
Recently, film-forming hydrogels (FFHs) have become considered an attractive means of transdermal delivery due to the potential advantages over other conventional products, including high flexibility, resistance to wearing off and sustained drug permeation [10]. FFHs are composed of hydrogel and film-forming agents in the presence of a volatile solvent that easily allows the material to transition from a hydrogel into a dry film after the application if the gel onto the skin surface rapid solvent evaporation [10, 11]. While films have the capacity to prolong drug release and resist detachment due to wear and washing through increased adhesion, they still have some limitations, such as inflexibility, limited shape and size, and the potential to cause skin irritation. In contrast, hydrogels are flexible; however, the frequent dosing and poor resistance to detachment due to wear and washing are inconvenient for patients. Hence, the application of FFHs could leverage the advantages of both hydrogels and films for more effective treatment. However, current efforts have focused on the direct loading of only pure drugs in film-forming gels. Additionally, these gels are not suitable for the delivery of poorly water-soluble drugs, which account for more than 40% of drugs with market approval and nearly 90% of molecules in the discovery pipeline [12] because the poor solubility will hamper their release at the target site, leading to ineffective treatment. To the best of our knowledge, an FFH embedded with nanoparticles has not yet been described in the literature. This study describes the fabrication of a film-forming nanogel (FFN) that quickly transforms from a gel into a versatile film containing drug nanoparticles for immediate treatment and protection regardless of the site of application on the skin, thus allowing prolonged release of the drug nanoparticles over a long period of time for improved treatment and protection. This work, which includes the development and detailed characterization of the film-forming gel incorporating nanoparticles, provides an understanding of the innovative FFN approach for the effective transdermal delivery of poorly water-soluble drugs.
2. Materials and methods
2.1. Materials
Curcumin was purchased from Merck Ltd. (Germany). Gelatine (gel strength, 225 g Bloom, type B), zein and glutaraldehyde (GTA) 25% solution were purchased from Sigma-Aldrich, Inc. (USA). Poloxamer 407 (POX 407) was obtained from BASF chemical company (Germany). HPMC 4000 was supplied by Dow Chemical Company (USA). Oleic acid, glycerol and absolute ethanol were obtained from Xilong Chemical Company (China). Methanol used for high-performance liquid chromatography (HPLC) analysis was purchased from Thermo Fisher Scientific (USA).
2.2. Methods
2.2.1. Preparation of FFH
HPMC 4000 was dispersed slowly in pre-heated water (60 °C) under continuous stirring to form a cloudy swelling solution. Then, the HPMC solution was cooled at −4 °C for 10 min. Simultaneously, zein, as a film-forming polymer, was dissolved in ethanol solution (90% v/v) at ambient temperature until a transparent solution appeared. The zein/HPMC FFH was obtained by blending zein and HPMC as film-forming and hydrogel agent, respectively, followed by gentle stirring for 2 h. After 2 h, a plasticizer or a mixture of plasticizers was added to the zein/HPMC blend and stirred thoroughly at room temperature. The final samples were stored in tubes and sealed tightly until further use. The composition of FFHs with varying concentrations of polymers and plasticizers is illustrated in table 1.
Table 1. FFH formulations (wt%).
| Plasticizer | |||||||
|---|---|---|---|---|---|---|---|
| Code | HPMC | Zein | Oleic acid | Glycerol | Ethanol | Water | Total |
| F1 | 3% | 3% | — | — | 64% | 30% | 100% |
| F2 | 3.5% | 2.5% | — | — | 64% | 30% | 100% |
| F3 | 4% | 2% | — | — | 64% | 30% | 100% |
| F4 | 3% | 3% | — | 1% | 63% | 30% | 100% |
| F5 | 3% | 3% | 1% | — | 63% | 30% | 100% |
| F6 | 3% | 3% | 0.5% | — | 63.5% | 30% | 100% |
| F7 | 3% | 3% | 1.5% | — | 62.5% | 30% | 100% |
2.2.2. Film-forming time
2.2.2.1. In vitro testing
The FFH was transferred onto a microscope slide (Duran, 76 × 26 mm) in a pre-determined area (10 mg cm−2) at 25 °C [9]. The film-forming process was observed visually, and the film-drying time was recorded.
2.2.2.2. Ex vivo testing
Porcine skin was preserved in 0.9% NaCl solution. The fat layer was removed to obtain an approximately 1 mm-thick sample of the top layers. Additionally, the skin was cut to a size of 1 × 1 cm2. The surface was washed with absolute ethanol to gain a dry substrate. Then, the skin was placed on the surface of a glass petri dish pre-heated at 37 °C. Subsequently, 10 mg of the FFH was spread on 1 cm2 of the skin sample, and the film-forming time on skin was measured.
2.2.3. Preparation of curcumin-loaded GNPs
CUR-GNPs were prepared by sonoprecipitation (table 2). First, 200 mg of gelatine was dissolved in 20 ml of water at 40 °C. Meanwhile, CUR and POX 407 were dissolved in 5 ml of methanol at room temperature until a transparent solution was obtained (4 mg ml−1 of CUR in the solvent phase). Then, the CUR/POX 407 mixture was dropped quickly into the dispersion phase under moderate magnetic stirring at 750 rpm. Then, the samples were immediately emulsified using an ultrasonic liquid processor (Qsonica, USA) with different sonication times and amplitudes. All samples were maintained in a water bath at 20 °C ± 1 °C during the sonication process to circumvent thermal variations. After ultrasonication, the samples were stirred at 30 °C during the dropwise addition of GTA 5% (v/v) solution to crosslink the particles. The methanol was completely evaporated after 2 h. The mixture was then centrifuged at 1000 rpm for 30 min to gradually remove small quantities of aggregated particles at the bottom of the tube, and the supernatant was lyophilized at −50 °C for 24 h to collect CUR-GNP powder.
Table 2. Curcumin-loaded GNP formulations.
| Sonication | ||||||
|---|---|---|---|---|---|---|
| Code | Gelatine (mg) | Curcumin (mg) | POX 407 (mg) | Amplitude | Time (min) | GTA (μl) |
| N1 | 200 | 20 | 250 | 20 | 10 | 30 |
| N2 | 200 | 20 | 250 | 30 | 10 | 30 |
| N3 | 200 | 20 | 250 | 40 | 10 | 30 |
| N4 | 150 | 20 | 250 | 30 | 10 | 30 |
| N5 | 200 | 20 | 375 | 30 | 10 | 30 |
| N6 | 200 | 20 | 200 | 30 | 10 | 30 |
| N7 | 200 | 20 | 250 | 30 | 20 | 30 |
| N8 | 200 | 20 | 250 | 30 | 30 | 30 |
| N9 | 200 | 20 | 250 | 30 | 20 | 50 |
2.2.4. Preparation of FFH containing CUR-GNPs
A pre-determined quantity of lyophilized CUR-GNPs (equivalent to 1 mg of CUR) was dispersed slowly in 1 g of FFH under magnetic stirring at 250 rpm for 24 h at ambient temperature to ensure that all of the nanoparticles were dispersed in the gel, forming a homogeneous mixture.
2.2.5. Characterization of CUR-GNPs
The particle size, polydispersity index (PDI), and zeta potential of the CUR-GNPs were analysed using a Zetasizer Nano Series system (Malvern Instrument Limited, UK). First, 20 μl of the CUR-GNP solution was diluted with 20 ml of methanol followed by vortexing prior to analysis. To determine the particle size of the CUR-GNPs in the FFH, 5 mg of FFN was dispersed in 10 ml of a complex solvent containing ethanol and distilled water corresponding to the ratio of the FFH preparation under stirring for 1 h. Measurements were performed in triplicate at 25 °C.
2.2.5.1. Entrapment efficiency (EE) and drug loading (DL)
CUR-GNPs (equivalent to 1 mg of CUR) were dispersed in 25 ml of distilled water followed by sonication for 30 min at 50 °C to extract CUR completely. Then, absolute ethanol was added to the mixture under shaking and cooled to room temperature. The final volume was adjusted accurately to obtain 50 ml. Then, 100 μl of the solution was diluted with 900 μl of methanol for HPLC analysis (as described in section 2.2.9). Measurements were performed in triplicate for each sample, and the quantity of CUR entrapped in the form of CUR-GNPs was calculated using the following equations:

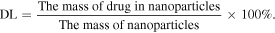
2.2.6. Permeation study
The in vitro drug release of CUR-GNPs from the FFH was determined using a Franz diffusion cell (area = 3.8 cm2, receptor chamber volume = 20 ml) with a cellulose diffusion membrane (Ø = 25 mm, 0.45 μm, Sartorius, Germany). The membrane was activated by drenching in isopropyl myristate (IPM) for 24 h before testing. Three Franz diffusion cells were equilibrated for 15 min in a water bath at 37 °C ± 0.5 °C before the sample was applied. The receptor medium, consisting of 20 ml of ethanol 20% (v/v), was continuously stirred at 500 rpm and 37 °C [13, 14]. The FFN (10 mg cm−2) was evenly spread on the surface of the membrane. Then, 500 μl of the receptor medium was collected after 1, 2, 4, 6, 8, 12, 18, and 24 h and replenished by an equivalent amount of fresh medium. For HPLC analysis, 100 μl of the solution was diluted with 900 μl of methanol.
2.2.7. HPLC analysis
The quantity of CUR was determined using an Ultimate 3000 HPLC system (Thermo Fisher Scientific, Inc., USA). HPLC analysis was performed using a reverse-phase column (150 × 4.6 mm, C18). The mobile phase consisted of methanol and water at a ratio of 80:20 (v/v), with a flow rate of 1.2 ml min−1. The run time and UV/Vis detector were set to 5 min and a wavelength of 425 nm.
2.2.8. Powder x-ray diffraction (PXRD)
The crystallinity of the dry films, pure CUR and carriers was determined using a Powder x-ray diffractometer (D2 Phaser, Bruker, Germany) with Cu radiation at 30 kV and 100 mA. All of the samples were scanned across diffraction angles (2θ) ranging from 10° to 50° with a step size of 0.02° and a rate of 2θ/s.
2.2.9. Fourier-transform infrared spectroscopy (FTIR)
An FTIR spectrophotometer (Vertex 70, Bruker, USA) was employed to obtain the spectra of dry films, pure CUR and carriers. One milligram of the sample was dispersed in 200 mg of dry potassium bromide (KBr). The compressed mixture of 1 mg of sample and 200 mg of KBr was scanned at wavelengths from 500 to 4000 cm−1 with a resolution of 4 cm−1.
2.2.10. Statistical analysis
All experiments were conducted three times. The data are presented as the mean ± SD and were analysed by ANOVA (SigmaPlot version 11). Statistically, P < 0.05 was considered to indicate a significant difference.
3. Results and discussion
3.1. Film-forming time
The natural characteristics and concentration of polymers could affect the drug delivery capability and mechanical properties of FFHs, such as the drying time, viscosity and visual appearance [15]. In our previous research, a zein/HPMC 4000 blend showed potential for application in modulating drug crystal changes and enhancing drug dissolution for solid dosage forms [16]. Furthermore, zein and HPMC are considered potential film-forming agents due to the manifestation of HPMC 4000 and zein as a hydrogel and film, respectively. In the FFH system, the concentration of polymeric film-forming agents is crucial. If it is too low, the FFH has low viscosity, which is not suitable for film formation. Meanwhile, the presence of a high polymer concentration results in a denser matrix that serves as a barrier to drug release and slows the film-forming time. Moreover, a high concentration of a hydrophobic polymer without plasticizers could result in brittle drying films. In this study, the concentration of polymeric film-forming agents was maintained at 6% of the total mass of the FFH. Additionally, different ratios of zein to HPMC 4000 were investigated to elucidate the mechanical properties of the FFH. As illustrated in figure 1 (top), an increase in the concentration of the HPMC 4000 solution as the hydrogel and a decrease in the concentration of zein as the film-forming agent did not significantly change the drying time of the FFH in vitro or ex vivo. However, an increase in the HPMC 4000 concentration resulted in a high viscosity and phase separation. Therefore, equal concentrations of zein and HPMC 4000 in the FFH (F1) were chosen for further investigation.
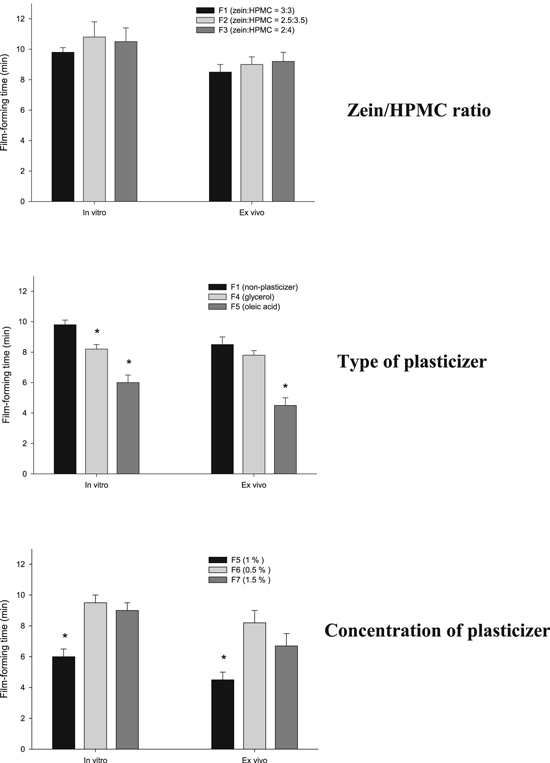
Figure 1. Effects of different compositions on the FFH film-forming time: zein/HPMC ratio (top); type of plasticizer (middle); concentration of plasticizer (bottom). The * indicates a significant difference in the film-forming time of the samples in each test.
Download figure:
Standard image High-resolution imagePlasticizers are low-molecular-weight substances that enhance the flexibility of polymers. Hence, the addition of a plasticizer could result in a flexible dry film that is less brittle due to a reduced glass transition temperature (Tg) and increased mobility of polymer chains in the matrix [17]. On the other hand, the nature and concentration of plasticizers also impact film formation [18, 19]. Therefore, different types and concentrations of plasticizers were studied, as illustrated in figure 1 (middle and bottom, respectively). As depicted in figure 1 (middle), the film-forming time of FFHs containing plasticizers showed an improvement, as the drying times were shorted than those of FFHs without plasticizers (P < 0.05). The presence of oleic acid resulted in a faster drying time than the presence of glycerol in the FFH (P < 0.05), which could be because glycerol is more hydrophilic than oleic acid and thereby prolonged the drying time of the FFH. In conclusion, the presence of a plasticizer resulted in a fast drying time, and oleic acid is a pertinent plasticizer for FFHs.
Figure 1 (bottom) demonstrates the drying times achieved by modifying the plasticizer concentration in the FFH F5. Obviously, the higher concentration of oleic acid in F7 resulted in a drying time longer than that of F5. This could be the result of the denser matrix inhibiting solvent evaporation. A lower plasticizer concentration also resulted in a longer film-forming time due to the reduced mobility of polymer chains in the FFH. In conclusion, the presence of 1% oleic acid in the FFH improved the film-forming time (P < 0.05), and the FFH system containing HPMC/zein/oleic acid/ethanol/water at a ratio of 3/3/1/63/30 in a given mass fraction (%) was considered optimal for loading drugs and CUR-GNPs.
3.2. Characterization of CUR-GNPs
3.2.1. Particle characteristics
Ultrasonication was employed to reduce the particle size by varying the intensity of the sonic waves to create mechanical stress in the samples, resulting in a uniform and stable dispersion. The profile in figure 2(A) shows that the particle size was less than 300 nm and that the particles had a moderate zeta potential of 30–35 mV. Increasing the amplitude from 20 to 30 resulted in the delivery of more energy to break down the samples; hence, the mean particle size also decreased from 279.9 nm to 242.1 nm. However, the application of the high amplitude of 40 resulted in a slightly increased particle size. Moreover, there was no significant difference in the average particle size between N2 and N3 (P > 0.05). This result could be explained by the high intensity of the sonic waves corresponding to the high amplitude, leading to the destruction of the nanoparticles; thus, the drug may have dissociated from the nanoparticles and accumulated in the anti-solvent phase to form larger particles. Therefore, an amplitude of 30 was chosen as a suitable ultrasonic condition for particle size reduction.
The results shown in figure 2(B) indicate that an increase or decrease in the concentration of the POX 407 emulsifier led to an increase in the mean particle size. Decreasing the emulsifier concentration to 8 mg ml−1 resulted in a less stable system and the formation of aggregates. Meanwhile, a high concentration of POX 407 resulted in the solubilization of CUR or gelatine in the precipitation process, which further reduced the stability of the system. Thus, an emulsifier concentration of 10 mg ml−1 was applied in further experiments.

Figure 2. Particle size and zeta potential of CUR-GNPs fabricated under different conditions: (A) level of sonication; (B) concentration of emulsifier; (C) gelatine concentration; (D) sonication time; (E) crosslinker concentration. In each figure, significant differences among the samples are indicated by the * symbol.
Download figure:
Standard image High-resolution imageOn the other hand, the gelatine concentration could only be increased to a limited extent due to the high viscosity of the anti-solvent phase, which further reduced the uniformity of the dispersion of the solvent phase. Moreover, increasing the gelatine concentration could lead to high concentration of free gelatine in the solution after nanoprecipitation. Consequently, the aggregation of this free gelatine resulted in the formation of larger particles through crosslinking, and it became more difficult to obtain a uniform dispersion of CUR-GNPs incorporated in the FFH, as gelatine is practically insoluble in most organic solvents and the FFH system contains ethanol. Hence, the concentration of gelatine in N4 should be reduced. However, decreasing the gelatine concentration of 7.5 mg ml−1 in N4 (figure 2(C)) resulted in a significantly increased particle size and decreased zeta potential compared to those of N2. It could be reasoned that a low gelatine concentration is not sufficient to encapsulate CUR/POX 407 inside nanoparticles.
Figure 2(D) depicts the particle size of N2, N7 and N8 resulting from ultrasonication at an amplitude of 30 for 10, 20, and 30 min, respectively. Compared with N2, the increased sonication time of N7 resulted in a significant increase in the zeta potential (P < 0.05). Moreover, there was no significant difference in particle size between N2 and N7 (P > 0.05); however, the encapsulation efficiency and drug loading of N2 were lower than those of N7 (details provided in section 3.2.2). In contrast to N7, when the sonication time was prolonged to 30 min for N8, the particle size was significantly increased to greater than 650 nm, and the zeta potential was reduced due to thermal variation resulting from the long period of sonication.
GTA 5% (v/v) solution was employed as a crosslinker to prevent the aggregation of and stabilize the CUR-GNPs. Different volumes of GTA were added dropwise. As shown in figure 2(E), the increased volume of GTA in N9 drastically increased the average particle size by approximately twofold compared to that of N7. Reasonably, greater concentrations of GTA resulted in the crosslinking of multiple groups and an increased degree of reticulation, thereby leading to an increased particle size. The use of less than 30 μl of GTA resulted in a large extent of aggregation because the quantity of crosslinker was insufficient to harden the nanoparticles (data not shown).
3.2.2. Encapsulation efficiency (EE) and percentage of drug loading (DL)
Based on the characterization of the CUR-GNPs, N1, N2 and N7 were selected for further investigation of the EE and DL (table 3) due to the smaller average particle size and the moderate zeta potential. The results show no significant difference in entrapment efficiency or drug loading between N1 and N2 (P > 0.05). Meanwhile, although there was no significant difference in particle size between N2 and N7, N7 performed significantly better, with the highest EE and DL of all other formulations (P < 0.05). Specifically, approximately 50% of the CUR was encapsulated in the nanoparticles, which were the smallest (233.4 nm) and showed good stability with the highest zeta potential (35.9 mV). Therefore, N7 was chosen as an optimal CUR-GNP model for dispersion in the FFH.
3.3. Characterization of CUR-GNP-loaded FFH
As illustrated in figure 3(A), the film-forming time of the FFH loaded with CUR-GNPs (F5-N7) was not significantly different from that of the FFH (F5) tested ex vivo (P > 0.05). Therefore, the presence of the CUR-GNPs did not considerably affect the drying time of the FFH system. In addition, the change in the average particle size of N7 after being loaded into F5 was negligible (figure 3(B)). Meanwhile, the magnitude of the zeta potential of N7 decreased nearly eightfold from 35.6 to −4.5 mV. This could be due to the high viscosity of the dispersion phase (FFH) and particle-particle interactions between the CUR-GNPs and FFH.
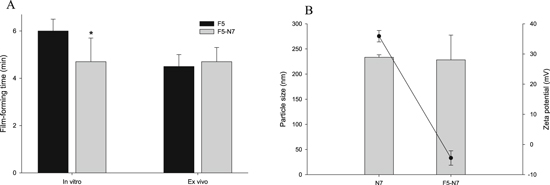
Figure 3. Characterization of CUR-GNP-loaded FFH: (A) film-forming time; (B) particle size and zeta potential.
Download figure:
Standard image High-resolution image3.4. Drug permeability of CUR-GNP-loaded FFH
Compared to the pure drug, the CUR-GNPs exhibited significantly improved diffusion (figure 4). The percentage of drug permeation from the FFH containing pure CUR was approximately 12% due to poor solubility, which prevented the drug from permeating through the membrane. Meanwhile, more than 60% of the CUR was released from the nanoparticles in F5-N7 after 12 h, and after 24 h, the percentage of drug permeation was 85%. Thus, the presence of the CUR-GNPs in the FFH led to improved drug release compared with the presence of pure CUR due to the smaller particle size.
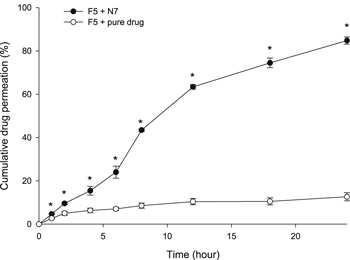
Figure 4. Drug permeability profile determined Franz diffusion cell testing. The * indicates a significant difference in drug permeability between F5-pure drug (CUR) and F5-N7 (CUR-NPs).
Download figure:
Standard image High-resolution image3.5. Physicochemical characterization of CUR-GNP-loaded FFH
PXRD analysis was performed to determine the crystallinity of CUR-GNPs, FFH loaded with CUR-GNPs, and pure CUR (figure 5). The diffractogram of pure CUR shows numerous peaks, indicating its natural high crystallinity. Meanwhile, the PXRD patterns of N7, F5-N7 and F5-CUR do not show the various characteristic peaks of CUR, which indicates a reduction in the crystallinity of the drug. Furthermore, the pattern of F5-N7 shows a reduced intensity for the two characteristic peaks of POX 407 at 19.4 and 23.2 2θ compared to those peaks in the pattern of N7, indicating that F5-N7 is more amorphous than N7. In contrast, the pattern of CUR-F5 exhibits a high-intensity, broad peak ranging from 13.3 to 21.3 2θ, which indicates that the release of drugs from a dry F5-CUR film would be further decreased.
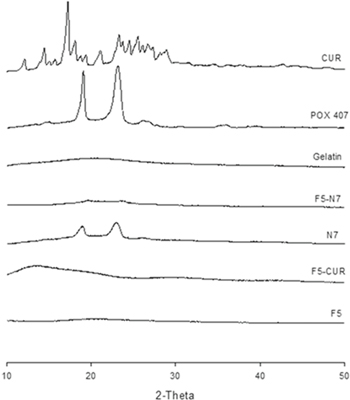
Figure 5. PXRD patterns of pure CUR, gelatine, POX 407, F5, N7, F5-N7 and F5-CUR.
Download figure:
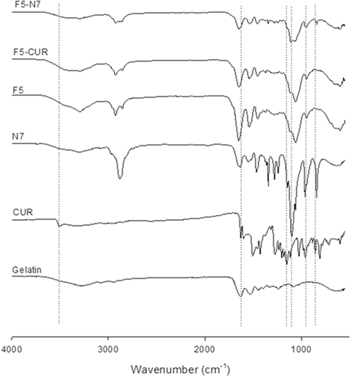
Standard image High-resolution imageFigure 6 shows the overlaid FTIR spectra of gelatine, CUR, N7, F5-CUR and F5-N7. A sharp peak at 3507.1 cm−1 and a broad peak at 3293 cm−1 were observed on the spectrum of CUR, indicating the presence of −OH groups [20]. Additionally, the characteristic peak at 1627 cm−1 could be predominantly attributed to C=O bonds [21]. Interestingly, the FTIR spectra of N7, F5-CUR and F5-N7 do not show the peak at 3507.1 cm−1 associated with −OH groups, which indicates the occurrence of hydrogen bonding between CUR and the carrier in each formulation. For N7, the C=O peak shifted from 1629.8 to 1648.6 cm−1, indicating interactions between F5 and N7 through the −COOH groups of gelatine and the –NH groups of zein; thus, the zeta potential of the CUR-GNPs was reduced in magnitude after the particles were loaded into the FFH. In conclusion, the characteristic peaks of CUR in the form of CUR-GNPs in the FFH remained unchanged compared to those of pure CUR in F5. Furthermore, even though the shifting peaks of the CUR-GNPs revealed a reduction in zeta potential, the presence of N7 in the FFH further enhanced the diffusion rate of CUR without any effects on the size of the CUR-GNPs.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. FTIR spectra of pure CUR, gelatine, POX 407, F5, N7, F5-N7 and F5-CUR.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusion
The FFN presented in the current study explored in terms of its potential for application in transdermal drug delivery, indicated by a fast film-forming time, uniform dispersion and sustained permeation of high drug concentrations for 24 h. Moreover, the presence of CUR-GNPs did not affect the film-forming time of FFH and enhanced the drug permeability. In conclusion, F5-N7 is an optimal formulation for achieving high drug permeability and fast drying in further studies of transdermal applications.
Acknowledgments
We would like to thank the International University for the support of our work. Dr Phuong HL Tran is the recipient of Australian Research Council's Discovery Early Career Researcher Award (Project Number DE160100900).