






Abstract
Phylogenetic reconstructions of transmission events from individuals with acute human immunodeficiency virus (HIV) infection are conducted to illustrate this group's heightened infectivity. Varied definitions of acute infection and assumptions about observed phylogenetic clusters may produce misleading results. We conducted a phylogenetic analysis of HIV pol sequences from 165 European patients with estimated infection dates and calculated the difference between dates within clusters. Nine phylogenetic clusters were observed. Comparison of dates within clusters revealed that only 2 could have been generated during acute infection. Previous analyses may have incorrectly assigned transmission events to the acutely HIV infected when they were more likely to have occurred during chronic infection.
HIV-infected individuals may be more likely to transmit their virus onward during acute infection than during chronic infection [1]. This is because acute infection is associated with elevated blood and genital fluid viral loads [2] at a time when infection is frequently undiagnosed. Individuals who are acutely infected with drug-resistant HIV strains may also disproportionately drive further transmission of drug-resistant viruses because of a combination of increased infectivity and the persistence of resistant viruses in their plasma [3]. To ensure that prevention initiatives (such as promotion of HIV testing and adherence to antiretroviral therapy) are effective, it is important to measure the extent to which patients with acute HIV infection generate transmission at the population level, including the transmission of drug-resistant strains.
Sequence-based antiretroviral resistance testing has become commonplace to inform treatment options [4]; this has resulted in an accumulation of HIV pol sequences, the variability of which is adequate for phylogenetic reconstruction of transmission events [5]. Phylogenetic reconstructions of transmission events that combine HIV pol sequences with data on patients' infection stage (obtained either by comparing the time interval between the date of the last negative HIV test result and diagnosis [6] or by laboratory serological algorithms [7]) have the potential to enhance our understanding of the role that the acutely HIV infected play in generating HIV transmission. Through such methods, explorative phylogenetic analyses have identified possible transmission events generated by the acutely HIV infected, illustrating the elevated transmission potential among this group [8]. More recently, Brenner et al. [9] attempted to measure the extent to which the acutely HIV infected generate new HIV transmission events by comparing the proportion of new transmissions generated by the acutely and chronically infected in turn.
However, interpretation and comparison between studies is difficult because of varied sampling strategies and definitions of acute HIV infection used. Importantly, the latter may serve to overestimate the force of transmission from this population. Viral load is known to peak less than a month after infection but remains elevated for ∼10 weeks [2]. However, definitions of acute infection and laboratory techniques used to identify it vary [7, 10, 11], and those frequently adopted mean that patients can be so categorized >6 months after infection [8, 9]. Moreover, studies do not consider the transient nature of acute infection: phylogenetic analyses frequently categorize patients as being acutely or chronically infected according to their infection stage at diagnosis [8, 9]. However, all patients experience acute and chronic infection, and the observation of possible transmission events (ascertained through phylogenetic reconstruction) between patients diagnosed during acute infection does not necessarily mean that transmission occurred while the patients were acutely HIV infected. For instance, Brenner et al. [9] showed that the mean interval between infection dates within clusters was 15 months-suggesting that most identified transmissions originated from chronically HIV-infected individuals- but interpreted these as acute-to-acute transmissions.
We conducted a phylogenetic analysis of a previously described HIV-infected European population [6] that used precise definitions of acute HIV infection to identify instances of possible transmission events, including transmission of drug-resistant strains. We calculated the difference between the estimated infection dates within the observed clusters to determine whether transmission could have occurred during acute infection. The analysis is not an attempt to measure the force of infection from the acutely HIV infected but a demonstration of the need for rigor in the design and interpretation of such analyses.
Methods. The study population was derived from CASCADE (http://www.cascade-collaboration.org) [12], an ongoing international collaboration that monitors events among HIV-infected individuals throughout their infections. HIV-1- infected patients were included provided they were >15 years old and had a reliable and precise date of infection. For the present analysis, this involved either an HIV-positive test result within 90 days of an HIV-negative test result or an HIV antibody-negative test result with polymerase chain reaction (PCR) positivity. The estimated infection date was taken as the midpoint between the diagnosis date and last negative test result for the former and as the date the sample was taken for the latter.
The first plasma sample available after the estimated HIV infection date was obtained for genotypic resistance testing. Samples obtained >18 months after the estimated infection date or from drug-experienced patients were excluded. Nucleotide sequence data spanned the protease gene and at least codons 41– 236 of reverse transcriptase (RT). The sequencing methodology has been described previously [6]. Drug-resistance mutations were defined on the basis of the standardized list of mutations for use in epidemiological studies [13].
Before phylogenetic analysis, the mutation sites associated with drug resistance [13] were deleted from all sequences. Protease and RT sequences were aligned across 998 nt and imported into the tree-building software PAUP (version 4.0b10). A neighbor-joining tree was created under the general time-reversible (GTR) model with gamma rate heterogeneity set at 0.5. The alignment was run through Modeltest software (version 3.7) to select the best-fitting evolutionary model. The GTR+I+G model was selected, and a heuristic search was conducted for a maximum-likelihood tree by means of this model and its derived parameters (proportion of invariable sites, 0.43; gamma distribution, 0.79), using the initial neighbor-joining tree as the starting tree. The robustness of the tree was evaluated by bootstrap analysis with 100 replicates. Genetic distances were calculated from the maximum-likelihood tree topology. Branches comprising 2 or more sequences with a bootstrap value of >99% and a genetic distance of <0.015 nucleotide substitutions per site were considered to constitute a cluster, representing possible transmission [5].To assess whether a possible transmission was generated by an individual during acute infection, clusters containing sequences with a difference of <180 days between estimated infection dates were interpreted as being a possible acute-to-acute transmission.
Ethics approval for cohorts contributing to the CASCADE Collaboration is maintained according to the respective national regulations in the countries concerned. The nucleotide sequences used in this study were deposited into GenBank under the accession numbers FJ030643-FJ030807.
Results. Of 8993 patients with an estimated infection date from data pooled in December 2004, HIV sequences were submitted from 10 European cohorts. Of these, 165 sequences were complete, were from drug-naive patients, and fulfilled the definition of acute HIV infection. Specifically, 131 (79%) had had a documented seronegative test result at least 90 days before their diagnosis (mean number of days between tests, 38 days), and 34 (21%) were HIV antibody negative with PCR positivity.
The majority of sequences were from men who had had sex with men (120/165 [73%]). The sample proportion from each country varied annually; overall, 23% (38/165) of the samples were from Italy, 23% (38/165) were from France, 20% (33/165) were from Germany, and 17% (28/165) were from the UK, with the remainder from Denmark, Spain, and the Netherlands.
Overall, 10% (17/165) of patients sampled were infected with a drug-resistant HIV-1 variant. Fourteen patients had a virus considered resistant to 1 or more nucleotide RT inhibitors (NRTIs), 3 carried a virus resistant to nonnucleotide RT inhibitors (NNRTIs), and 1 had a virus resistant to protease inhibitors. One patient carried virus with both NRTI and NNRTI mutations.
Of the 165 sequences, 18 (11%) formed a phylogenetic cluster with at least 1 other sequence (clusters A-I) (figure 1). These formed 9 phylogenetic clusters, with 2 sequences in each cluster. All 9 phylogenetic clusters were composed of sequences derived from patients sampled within the same country. Two of the 9 clusters contained sequences that had a difference between infection dates of <180 days (104 and 29 days for clusters E and G, respectively) (table 1). For the remaining 7 clusters, the average difference in infection dates within clusters was 675 days (range, 187–1571 days).

Phylogenetic tree showing 9 clusters and sequences with drug-resistance mutations within a European cohort of HIV-infected patients with well-estimated infection dates (<1995–2004). Phylogenetic clusters are shown in boldface and are labeled A-I; red diamonds denote drug-resistance mutations.
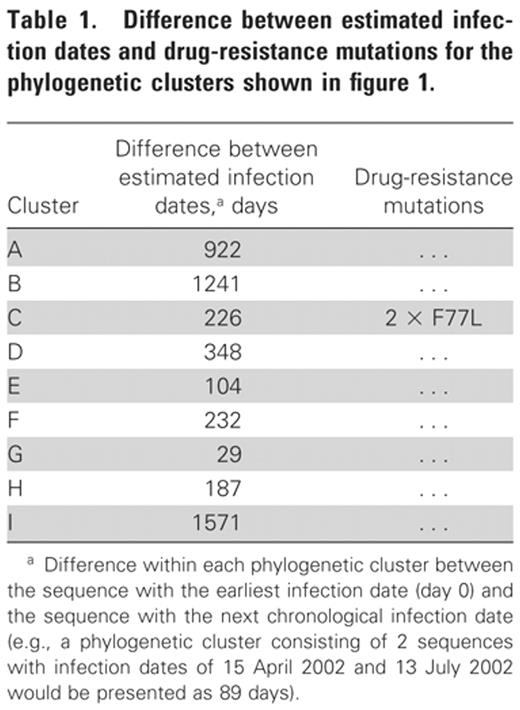
Of the 18 sequences with drug-resistance mutations, 2 formed a phylogenetic cluster (C) (figure 1). Both sequences shared the F77L mutation and were derived from patients with a difference of 226 days between their infection dates table 1.
Discussion. We conducted a phylogenetic analysis of HIV infected European patients with well-estimated infection dates and calculated the difference between infection dates within the possible transmission clusters identified. Nine transmissions may have occurred between individuals in this sample, including 1 possible instance of transmitted drug resistance. Of the possible transmissions events identified, only 2 could have been generated during acute HIV infection.
This analysis demonstrates the need for caution in the design and interpretation of phylogenetic analyses that link HIV pol sequences to data on patients' infection stage. For the first time, the time intervals between infection dates between patients involved in a possible transmission event have been explored. This comparison revealed that only 2 possible transmissions could have occurred during acute infection. The remainder are more likely to be transmissions from individuals with chronic infection. Previous analyses that use liberal definitions of acute HIV infection and do not take into account the transient nature of infection stage may inaccurately calculate the force of infection from this group, including the transmission of drug-resistant viruses [9], within the populations studied.
We recommend that future analyses should comprise sequences from patients from a local HIV-infected population and contain a large proportion of patients with well-estimated infection dates. Methods should then allow each patient's infection stage to change from acute to chronic to reflect the natural progression of HIV infection. Consequently, both acute and chronically HIV-infected patients will be represented in the sample. Additionally, we suggest that identified transmission events that involve patients with acute infection could be used to approximate the transmission date (i.e., transmissions involving an acutely HIV-infected individual are likely to have occurred around that patient's time of diagnosis). This method of dating will allow researchers to ascertain whether the patient most likely to have generated the acute infection was acutely or chronically infected at the transmission date (provided he or she also has an estimated infection date).
Our analysis is not a fresh attempt to measure the force of transmission from patients with acute HIV infection. The data were selected because they contained well-estimated infection dates. However, the size and the inconsistent geographic and demographic makeup of the population sample prohibits the extrapolation of results to the HIV-infected population and the calculation of the rate of transmission from the acutely HIVinfected population.
Further methodological challenges are applicable to all phylogenetic analyses of HIV-infected patients. First, although the bootstrapping and genetic distance cutoffs used are conservative and have been shown to reduce the proportion of false-positive clusters among an analysis of UK HIV sequences [5], it is not possible to conclusively demonstrate that the possible transmission events identified in the present analysis represent actual transmissions [14]. The clusters observed could each have been generated by a third party (not sampled) infecting each individual sequenced in the cluster or acting as an intermediary between the sequenced persons. Similarly, transmissions from the patients represented in this study would not be identified if the sequence from the infection generated was not included in the sample. Additionally, such studies cannot include sequences from the population with undiagnosed infections (a higher proportion of the acutely HIV infected are likely to have undiagnosed infections compared with those with chronic infection). Finally, the phylogenetic transmissions observed may be relative to the genetic diversity among the sampled sequences [14] rather than reflect absolute transmission events. For instance, Brenner et al. [9] found that the phylogenetic clusters observed among the acutely HIV infected were disrupted when additional sequences were added.
We have demonstrated the need for precise definitions of acute HIV infection in phylogenetic transmission reconstructions and the requirement that the possible transmission events observed through such analyses take into account the transient nature of acute infection. Future analyses should (1) use population data that contain a large proportion of patients with wellestimated infection dates, (2) allow each patient to progress from acute to chronic infection, (3) use transmissions involving acute infections to approximate the date of transmission and thereby ascertain the infection stage of the most likely transmitter at around the time of transmission, and (4) use a sample that is broadly representative of a local HIV-infected population with respect to geography and time. Only then can we attempt to measure with any precision the extent to which the acutely HIV infected drive HIV transmission at the population level.
CASCADE Collaboration.Steering Committee: Julia Del Amo (chair), Laurence Meyer (vice-chair), Heiner Bucher, Geneviève Chêne, Deenan Pillay, Maria Prins, Magda Rosinska, Caroline Sabin, and Giota Touloumi. Coordinating Centre: Kholoud Porter (project leader), Sara Lodi, Sarah Walker, Abdel Babiker, and Janet Darbyshire. Clinical Advisory Board: Heiner Bucher, Andrea de Luca, Martin Fisher, and Roberto Muga. Collaborators: Australia-Sydney AIDS Prospective Study and Sydney Primary HIV Infection Cohort (John Kaldor, Tony Kelleher, Tim Ramacciotti, Linda Gelgor, David Cooper, and Don Smith); Canada-South Alberta Clinic (John Gill); Denmark-Copenhagen HIV Seroconverter Cohort (Louise Bruun Jorgensen, Claus Nielsen, and Court Pedersen); Estonia-Tartu Ülikool (Irja Lutsar); France-Aquitaine Cohort (Geneviève Chêne, Francois Dabis, Rodolphe Thiebaut, and Bernard Masquelier), French Hospital Database (Dominique Costagliola and Marguerite Guiguet), Lyon Primary Infection Cohort (Philippe Vanhems), and SEROCO Cohort (Laurence Meyer and Faroudy Boufassa); Germany-German Cohort (Osamah Hamouda and Claudia Kucherer); Greece-Greek Haemophilia Cohort (Giota Touloumi, Nikos Pantazis, Angelos Hatzakis, Dimitrios Paraskevis, and Anastasia Karafoulidou); Italy-Italian Seroconversion Study (Giovanni Rezza, Maria Dorrucci, Benedetta Longo, and Claudia Balotta); Netherlands-Amsterdam Cohort Studies among Homosexual Men and Drug Users (Maria Prins, Liselotte van Asten, Akke van der Bij, Ronald Geskus, and Roel Coutinho); Norway-Oslo and Ulleval Hospital Cohorts (Mette Sannes, Oddbjorn Brubakk, Anne Eskild, and Johan N. Bruun); Poland-National Institute of Hygiene (Magdalena Rosinska); Portugal-Universidade Nova de Lisboa (Ricardo Camacho); Russia-Pasteur Institute (Tatyana Smolskaya); Spain-Badalona IDU Hospital Cohort (Roberto Muga), Barcelona IDU Cohort (Patricia Garcia de Olalla), Madrid Cohort (Julia Del Amo and Jorge del Romero), Valencia IDU Cohort (Santiago Pèrez- Hoyos and Ildefonso Hernandez Aguado); Switzerland-Swiss HIV Cohort (Heiner Bucher, Martin Rickenbach, and Patrick Francioli); Ukraine-Perinatal Prevention of AIDS Initiative (Ruslan Malyuta); United Kingdom-Edinburgh Hospital Cohort (Ray Brettle), Health Protection Agency (Valerie Delpech, Sam Lattimore, Gary Murphy, John Parry, and Noel Gill), Royal Free Haemophilia Cohort (Caroline Sabin and Christine Lee), UK Register of HIV Seroconverters (Kholoud Porter, Anne Johnson, Andrew Phillips, Abdel Babiker, Janet Darbyshire, and Valerie Delpech), University College London (Deenan Pillay), and University of Oxford (Harold Jaffe).
References
Potential conflicts of interest: none reported
Financial support: University College London Hospitals/University College London Comprehensive Biomedical Centre (support to this study). The CASCADE Collaboration has been funded through the European Union (grants BMH4-CT97-2550, QLK2-2000-01431, QLRT- 2001- 01708, and LSHP-CT-2006-018949).
Study group members are listed after the text.


{kind=link}
{kind=link}