






Abstract
Lactoferrin is a glycoprotein present in most human mucosal secretions, including human milk. Lactoferrin is bacteriostatic in low iron media and, in some settings, bactericidal. Lactoferrin impairs ability of Shigella flexneri serotype 5 strain M90T to invade HeLa cells. To determine the mechanism by which lactoferrin decreases invasiveness of Shigella organisms, its effect on the major virulence proteins responsible for bacterial uptake by host cells was evaluated. Lactoferrin induced degradation of invasion plasmid antigens IpaB and, to a lesser extent, IpaC, the key proteins responsible for bacteria-directed phagocytosis by mammalian cells. The lipid A–binding N-terminal portion of lactoferrin (residues 1–33) induces release of invasion antigens but does not induce degradation of IpaBC. Lactoferrin does not directly degrade previously released invasion plasmid antigens but works by making IpaBC susceptible to breakdown by surface-expressed protease(s)
Although bacillary dysentery due to Shigella species is among the most communicable and severe forms of bacterial gastroenteritis in humans, it is uncommon in the first year of life. A prospective study of infants whose serum samples were cultured weekly from birth showed that asymptomatic colonization with Shigella species is frequent [1]. Epidemiologic studies have repeatedly demonstrated that breast-feeding decreases the severity of Shigella infection in infants who become colonized early in life [2–5]. Both immune and nonimmune components of milk may be relevant to this protection. Lactoferrin is a major nonimmune milk factor that has been suggested to be important in protecting infants from intestinal infections; it is an iron-binding 78-kDa glycoprotein that is resistant to proteolytic enzymes [6]. Lactoferrin is produced not only in breast milk but also in other mucosal secretions and phagocytic cells
Lactoferrin may protect against gram-negative bacteria in a variety of ways. It was originally thought that it impaired bacterial multiplication due to its ability to decrease availability of iron required for growth [7–13]. However, the antibacterial activity of lactoferrin is not due solely to its bacteriostatic iron-binding capacity [14, 15]. A pepsin-derived fragment of lactoferrin has iron-independent bactericidal activity that is associated with release of lipopolysaccharide (LPS) [16]. Lactoferrin kills or slows bacterial growth synergistically with other factors that may be present in mucosal secretions, including immunoglobulins [17], complement [18], and lysozyme [19]. Although lactoferrin binds Shigella porins [20], the precise role and relevance of lactoferrin in protection against shigellosis have not been fully defined
The fundamental virulence attribute of Shigella species is its ability to invade mammalian cells [21, 22]. Invasion plasmid antigens (IpaB and IpaC in particular) are responsible for this phenotype. The invasion plasmid antigens are secreted proteins rather than integral membrane proteins; they can be purified by the water extraction procedure described in Oaks et al. [23]. We found previously that lactoferrin impairs virulence of penta start-second-page>Shigella flexneri as manifested by decreased ability to invade HeLa cells [24]. The objective of the present studies was to determine the mechanism by which human lactoferrin interferes with Shigella invasiveness
Materials and Methods
Reagents and lactoferrin incubation procedure We used recombinant human lactoferrin (Agennix), prepared according to the method described in Ward et al. [25]. The preparation is 11% iron saturated. The concentration of lactoferrin used in all experiments was in the physiologic range; in human colostrum and milk, lactoferrin concentrations are ∼10 mg/mL (0.125 mM) and 1 mg/mL (0.0125 mM), respectively. The N-terminal 33-aa peptide of lactoferrin (SynPep) also was evaluated at the 0.125 mM concentration. The peptide was amidated at the C-terminal end to remove the charge associated with the free-carboxyl group. The peptide was purified by high-performance liquid chromatography, and purity was confirmed by mass spectroscopy. For all studies described, S. flexneri 5 strain M90T, previously stored in glycerol at −70°C, was grown overnight at 37°C on Congo Red agar to verify the presence of virulence genes. Strain M90T then was grown to log phase in brain-heart infusion broth, washed, and incubated with end-over-end rotation at 37°C for 60 min in 10 mM PBS (pH 7.4), in the absence or presence of lactoferrin
HeLa cell assays HeLa cells were grown in 24-well tissue culture plates at a concentration of 4×105 cells/well. For studies of bacterial adherence, strain M90T was labeled with tritiated thymidine (25 Ci/mmol; 1 μCi/mL of growth medium) and then washed to remove free label, incubated with lactoferrin or buffer for 1 h at 37°C, as described above, washed, added to HeLa cells at a 100:1 bacteria-to-target cell ratio, incubated for 15 min with HeLa cells, washed, and lysed with 1 N KOH. We measured the number of counts per minute (cpm) in a scintillation counter to determine the number of HeLa cell–associated bacteria. We calculated the number of bacteria attached per HeLa cell by determining the cpm per colony-forming unit and the number of HeLa cells
Growth measurement To determine whether lactoferrin affected growth, a log-phase growth of S. flexneri was inoculated into Luria-Bertani (LB) medium containing lactoferrin, transferrin (Sigma), bovine serum albumin (BSA; Sigma), equine skeletal myoglobin (Sigma), or PBS. Each protein was at a concentration of 1 mg/mL in 10 mM (pH 7.4) sodium phosphate buffer. We measured the optical density at 600 nm every 15 min for 75 min during log-phase growth of a culture without agitation at 37°C. Data are expressed as doubling time±SEM. We also determined growth curves in the presence of varying lactoferrin concentrations (0–0.125 mM)
Assays for lactoferrin-induced release of virulence antigens Log-phase S. flexneri were preincubated with or without various concentrations of lactoferrin for 1 h at 37°C. After centrifugation of the incubation mix, the bacterial proteins that had been released into the buffer were concentrated 10-fold by ultrafiltration (10-kDa cutoff) prior to electrophoresis and immunoblotting [26]. Antigens separated by 7.5% SDS-PAGE were transferred to nitrocellulose and reacted either with invasion plasmid antigen-specific human convalescent serum, with mouse Ipa-specific monoclonal antibodies (MAbs) or in the case of the intracellular spread, protein IcsA, with a rabbit polyclonal antibody to purified IcsA. Ipa-specific serum samples were prepared from convalescent serum samples from patients who had recently recovered from Shigella sonnei dysentery
Serum samples were preincubated with lactoferrin and with a sonicate of S. flexneri 5 M90T-A2, an avirulent noninvasive strain lacking the invasion plasmid antigens needed to remove antibodies to chromosomally encoded antigens. For some assays depending on the issue being addressed, we used serum that reacted with IpaB and IpaC; for other immunoblots, we used serum that reacted with all of the virulence antigens. We used a water extract preparation that contained the invasion plasmid antigens to demonstrate antibody specificity and the effect of lactoferrin on virulence antigens [23]. To confirm specificity, MAbs to IpaB (1H4), IpaC (2G2), and IpaD (16F8) [27, 28] and polyclonal rabbit antiserum to IcsA (rabbit 59; provided by E. V. Oaks, Department of Enteric Infections, Walter Reed Army Institute of Research, Washington, D.C.) were used. The anti–Ipa antibodies were detected with peroxidase-conjugated goat antiserum (either anti–human IgG, anti–mouse IgG, or anti–rabbit IgG; Sigma). For experiments in which the effect of iron saturation was determined, lactoferrin was saturated with ferric chloride in a molar ratio of 2:1 (2 ferric ions:1 lactoferrin molecule) several days prior to assays that used this preparation
To determine whether lactoferrin’s effect was on invasion plasmid antigens at the bacterial cell surface or on the proteins after they had already been released into the supernatant, lactoferrin was incubated for 1 h at 37°C with a previously prepared filter-sterilized invasion plasmid antigen preparation [23]. The reaction mix was then separated by 7.5% SDS-PAGE and transferred to nitrocellulose. An immunoblot was prepared by using absorbed polyclonal convalescent serum that reacted with all invasion plasmid antigens
Protease inhibitor treatment We incubated S. flexneri for 1 h at 37°C, as described above, with 0.125 mM lactoferrin in the presence or absence of compounds known to inhibit serine, cysteine, aspartate, and metalloproteases. The amounts used were the maximum concentrations suggested by the manufacturer (Sigma): AEBSF [4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride] (1 mM), 6-aminohexanoic acid (38 mM), antipain hydrochloride (100 uM), aprotinin (800 nM), benzamidine hydrochloride hydrate (4 mM), bestatin hydrochloride (40 uM), chymostatin (100 uM), E64 [trans-epoxysuccinyl-l-leucylamido-(4-guanidino)butane] (10 uM), EDTA (1 mM), N-ethylmaleimide (NEM; 1 mM), leupeptin hemisulfate (100 uM), pepstatin A (150 uM), phosphoramidon disodium salt (10 uM), and soybean trypsin inhibitor type I-S (5 mM). The released/degraded invasion plasmid antigens were detected by means of Western blot
Statistical methods Parametric data are presented as mean±SEM. The significance of differences between lactoferrin-treated and controls in invasion and adherence was determined by Student’s t test. Generation time data were analyzed by analysis of variance (ANOVA) and by comparison of regression lines
Results
There are several possible explanations for the previously described decrease in intracellular bacteria in a HeLa invasion model after a very brief exposure to lactoferrin. [24]. Such a decrease could reflect lactoferrin-mediated impaired adherence of bacteria to HeLa cells. An alternate possibility is that the decrease in bacterial numbers in invasion assays could reflect impaired growth of S. flexneri since lactoferrin has a bacteriostatic effect on some organisms. A third possible explanation is that lactoferrin disrupts the internalization process. We evaluated each of these possibilities
Lactoferrin does not affect ability of S. flexneri to adhere to HeLa cells Bacterial adherence, assessed by using 3H-labeled S. flexneri briefly incubated with HeLa cells, was not affected by whether the bacteria had been preincubated with buffer alone or lactoferrin in buffer. The number of bacteria attached to each HeLa cell was essentially identical after either pretreatment (13±10 vs. 13±8 bacteria/HeLa cell in the absence or presence of lactoferrin [0.125 mM], respectively; triplicate assays done 3 times)
Growth measurements Growth of lactoferrin-treated organisms was determined with lactoferrin and several different proteins added to LB medium as controls. The generation time in LB broth was not affected by lactoferrin, BSA, equine skeletal myoglobin, human transferrin, or sodium phosphate buffer. The doubling time of M90T grown in unshaken media with myoglobin (63.6±1.2 min), transferrin (62.6±2.4 min), albumin (58.9±3.1 min), lactoferrin (63.4±2.2 min), and sodium phosphate buffer (60.8 ± 2.0 min) was not significantly different by ANOVA. Growth rate was also unaffected by vary-ing the concentration of lactoferrin (0–0.125 mM) present in the growth medium (figure 1). Additional experiments in which organisms were preincubated with lactoferrin for 1 h, washed, and cultured in broth likewise showed no effect on subsequent growth. Thus, the internalization process became the focus of further studies
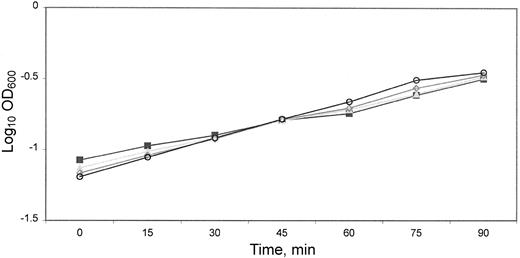
Growth of Shigella flexneri in the presence of lactoferrin: log-phase growth of M90T in Luria-Bertani medium containing lactoferrin at various concentrations (○, none; ▪, ▴, ◊, 0.125, 0.062, and 0.012 mM respectively). There was no difference in growth rate related to presence or absence of lactoferrin by comparison of the log-transformed linear regression lines
Lactoferrin causes release and degradation of invasion plasmid antigens Because lactoferrin interacts with multiple bacterial surface components, we hypothesized that it might disrupt the surface and cause loss of important virulence factors. Three virulence proteins, IpaB, IpaC, and IpaD, are critically important for initiating entry into HeLa cells [22]. IpaB and IpaD are involved in regulation of secretion while IpaB and IpaC form a complex on secretion, a complex sufficient to cause uptake and internalization [21, 29,–31]. Mutants that lack IpaA are not invasive so it is not a candidate for the observed lactoferrin effects
The Shigella supernatant treated with various concentrations of lactoferrin was examined by immunoblot to determine whether invasion plasmid antigens had been released; such loss of invasion antigens could account for the previously observed [24] decreased ability to invade HeLa cells. Immunoblots were prepared by using polyclonal convalescent human antiserum that had been preabsorbed with an avirulent S. flexneri mutant lacking the virulence plasmid so that only invasion plasmid antigens were detected. Absorbed antiserum failed to detect either lactoferrin or the avirulent S. flexneri lacking virulence plasmid antigens, strain M90T-A2 (not shown). Immunoblots showed that virulence antigens were lost into the supernatant (figures 2 and 3). The multiple bands between IpaB and IpaC suggested that IpaB had been released and degraded. In contrast to the release of antigens that occurs during the water extraction procedure, loss of invasion antigens in the presence of lactoferrin was associated with their degradation. Iron saturation had no effect on this loss/degradation of IpaB (figure 2). The iron-binding properties of lactoferrin are unrelated to its effect on invasion antigens
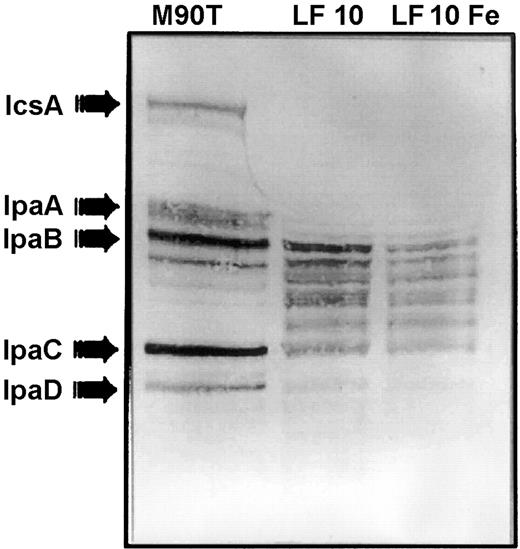
Effect of iron saturation on lactoferrin-mediated invasion plasmid antigen release. Water extract of Shigella flexneri (left lane)shows that the convalescent serum recognized the major invasion plasmid antigens released by the organism during the water extraction procedure. Incubation of S. flexneri with lactoferrin at 10 mg/mL (0.125 mM) for 1 h at 37°C was associated with loss and degradation of invasion plasmid antigens (lane LF 10).When lactoferrin was saturated with FeCl3 before incubation with bacteria, the pattern of loss/degradation of invasion antigens was unchanged (lane LF 10 Fe)
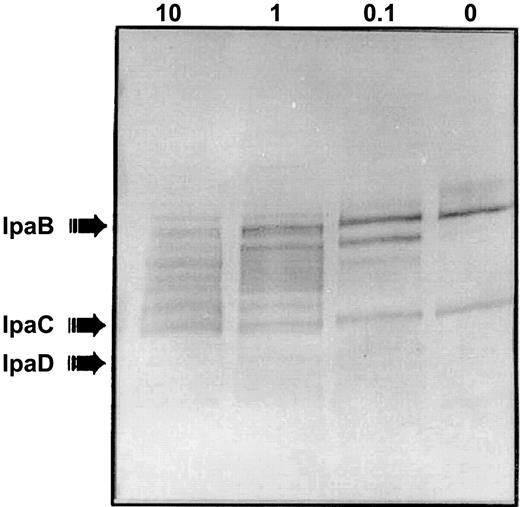
Immunoblot showing lactoferrin-mediated invasion plasmid antigen release is concentration dependent. When Shigella flexneri M90T was treated with a range of lactoferrin concentrations (10, 1, 0.1, and 0 mg/mL for 1 h at 37°C), a preabsorbed convalescent human serum sample reacted in a concentration-dependent fashion with invasion plasmid antigen protein that had been released into the supernatant. This blot used serum that reacted with IpaB and IpaC to demonstrate the effect of varying concentrations of lactoferrin. IpaB was released and degraded. Bands faintly visible suggested that IpaC also was broken down
The effect of lactoferrin on degradation of invasion plasmid antigens is concentration related. In the presence of only buffer, small amounts of IpaB and IpaC were released into the supernatant even with no lactoferrin present (figure 3, lane 0). Incubation with irrelevant proteins, BSA, and transferrin at the same molar concentration as lactoferrin gave a pattern identical to that seen with buffer alone in figure 3 (immunoblot not shown). The amount of IpaB released and degraded related directly to the amount of lactoferrin with which the S. flexneri was incubated: as intact IpaB disappeared at increasing concentrations of lactoferrin, breakdown products of IpaB appeared. Degradation could be due to a direct effect of lactoferrin on IpaB or could relate to the inherent instability of IpaB when not complexed with IpaC. This convalescent absorbed antiserum reacted faintly with multiple bands smaller than IpaC in the supernatant of lactoferrin-treated bacteria, suggesting the possibility that small amounts of IpaC might also be lost and degraded. However, IpaB was clearly lost far in excess of IpaC
To confirm these observations, we used MAbs in immuno-blots. We had anticipated that the human serum samples might better detect breakdown products than the MAbs if some critical epitope was lost; however, this was not the case. Immunoblots reacted with MAbs confirmed loss and breakdown of IpaB; however, with prolonged development of blots we found that IpaC, whose breakdown products had been poorly visualized with use of convalescent serum, was clearly lost and degraded along with IpaB. For both IpaB and IpaC, the amount of protein lost and degraded increased in a lactoferrin concentration-dependent fashion (figure 4, lanes 1 and 0.1)
![Immunoblot confirmation of IpaB and IpaC release by use of monoclonal antibodies (MAbs). MAbs were reacted with invasion plasmid antigens (lane WE [water extract]) and with the supernatant of Shigella flexneri M90T pretreated with lactoferrin (LF; 0, 0.1, and 1 mg/mL). MAb 1H4 (to IpaB) and MAb 2G2 (to IpaC) demonstrated a concentration-dependent release and degradation of IpaB and IpaC. MAb 2G2 demonstrated a breakdown of IpaC that was only faintly seen with the convalescent serum (see figure 3). IpaD and IcsA, in contrast, were not released into the supernatant with lactoferrin treatment. Composite figure is of 4 immunoblots; each reacted with a different MAb](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/187/1/10.1086_345875/2/m_187-1-87-fig004.jpeg?Expires=1716321378&Signature=TUcT~Qi8vUhOVCGZpcECRgJxXnG6x2v93KCkrAEiL~So4KCied6aAgqWy7o4OhomGCIpJPwU4UZG4rVLPzvQxHO95xIzyQz8m70jyXvERraDdxO0tXMbTx38~8SUbUvupoIWmWKqKKmYKBel1DuT7PabIY5wPDygnja-H9DGlBg93jmfceYgjp1yy-6n0i9yefZn2FODj5qkSlQ2Fuubv9To2GXBgFsTMnPbJIHUe1x7I~XGFvg3~jzZrf2eqNfY7dvlDhFAXX3bRf0n4HcP~4c49M4OIB1WKfj3iKSlevcgBfBBr518sQEg-92WS6NTyJV9qDtSzYLwOc3P2IfAlw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Immunoblot confirmation of IpaB and IpaC release by use of monoclonal antibodies (MAbs). MAbs were reacted with invasion plasmid antigens (lane WE [water extract]) and with the supernatant of Shigella flexneri M90T pretreated with lactoferrin (LF; 0, 0.1, and 1 mg/mL). MAb 1H4 (to IpaB) and MAb 2G2 (to IpaC) demonstrated a concentration-dependent release and degradation of IpaB and IpaC. MAb 2G2 demonstrated a breakdown of IpaC that was only faintly seen with the convalescent serum (see figure 3). IpaD and IcsA, in contrast, were not released into the supernatant with lactoferrin treatment. Composite figure is of 4 immunoblots; each reacted with a different MAb
Lactoferrin does not seem to induce secretion through hydrophobic interactions in a fashion analogous to Congo Red and other dyes [32]. Such dyes cause secretion of IpaD, IpaB, and IpaC. Loss of IpaD and IcsA into the supernatant was not detected after lactoferrin treatment, as indicated by studies with polyclonal antibodies and MAbs (figures 2 and 4). In the report of Bahrani et al. [32], immunoblots showed no evidence of degradation of invasion plasmid antigens after Congo Red–induced secretion. The findings in the present study did not reflect lactoferrin-mediated cell lysis. A silver stain of SDS-PAGE–separated supernatant of lactoferrin-treated bacteria showed only bands consistent with lactoferrin, IpaB, IpaC, and breakdown products of IpaB and/or IpaC. A whole-cell bacterial lysate had, as expected, innumerable bands on SDS-PAGE (not shown)
To investigate the role of the lipid A–binding N-terminal cationic portion of lactoferrin on release of virulence antigens, organisms were treated as above with a synthetic peptide consisting of aa 1–33, and release of proteins into the supernatant was evaluated. The N-terminal peptide (figure 5, lane peptide) did not cause degradation of Shigella virulence antigens like that seen with lactoferrin (figure 5, lane LF 10). Incubation with this peptide for 1 h did not interfere with lactoferrin subsequently triggering degradation of invasion antigens (figure 5, lanePeptide + LF 10). Peptide preparations synthesized independently by 2 manufacturers gave identical results in these studies. Studies with another lipid A–binding polypeptide, poly-myxin B (20,000 U/mL for 1 h at 37°C), caused neither loss/degradation of invasion plasmid antigens nor blockage of subsequent lactoferrin effect (not shown)
![The N-terminal 33-aa peptide of lactoferrin does not cause release and degradation of IpaB and IpaC. Shigella flexneri was incubated for 1 h at 37°C with lactoferrin (lane LF; 10 mg/mL [0.125 mM]), the N-terminal 33-aa peptide of lactoferrin (lane Peptide; 0.125 mM), or the phosphate buffer (lane PB) in which the proteins were dissolved. Released antigens were detected by Western blot in human convalescent serum that reacted with IpaB–IpaD in addition to several small undefined proteins. When lactoferrin (0.125 mM 37°C, 1-h incubation) was added to Shigella organisms treated for 1 h with the N-terminal 1–33 peptide, the additional treatment led to release and degradation of invasion antigens in a pattern similar to lactoferrin treatment alone](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/jid/187/1/10.1086_345875/2/m_187-1-87-fig005.jpeg?Expires=1716321378&Signature=yAg53oe9ggdkhX6GjMSLiBGE4k8LNGXfVKU7ZxnMJhT70w5g21LOWWFZGTcmoyTzCOEDZgo-ivg-UNgf6rKffNelDjByk68o8blr2Ign7Bz6dvtmYmE1VUnvFRl5nABn0e-bA190RVMZFWGHuOX5cb3wLF4RoQhT158mgagoyV1pz-uO8~nfnO6p83NuvO5XOJE1Qrp1ZxLrTc0lDwjlrxXyJA6GwfWSr6~5E0aYR6dSGJMITrlZTHKnttEobjfosl1D~Hm8eLSQiup977l-nm4dPS0r9MV7WZNpeA6a84vMmFQ5xtRsXzRO-LM0vrjjJKwZvjTtNtsI6p4YXNWmPw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The N-terminal 33-aa peptide of lactoferrin does not cause release and degradation of IpaB and IpaC. Shigella flexneri was incubated for 1 h at 37°C with lactoferrin (lane LF; 10 mg/mL [0.125 mM]), the N-terminal 33-aa peptide of lactoferrin (lane Peptide; 0.125 mM), or the phosphate buffer (lane PB) in which the proteins were dissolved. Released antigens were detected by Western blot in human convalescent serum that reacted with IpaB–IpaD in addition to several small undefined proteins. When lactoferrin (0.125 mM 37°C, 1-h incubation) was added to Shigella organisms treated for 1 h with the N-terminal 1–33 peptide, the additional treatment led to release and degradation of invasion antigens in a pattern similar to lactoferrin treatment alone
Protease inhibitors partially block lactoferrin-induced degradation of invasion antigens IpaB and IpaC Some serine protease inhibitors (antipain, chymostatin, and soybean trypsin inhibitor) blocked degradation of IpaC and partially blocked degradation of IpaB (figure 6). The Western blot shown demonstrates that benzamidine, EDTA, E-64, and pepstatin did not block lactoferrin-induced IpaBC degradation. Likewise, AEBSF, 6 aminohexanoic acid, bestatin, NEM, leupeptin, and phosphoramidon failed to block lactoferrin-induced degradation (not shown). The fact that soybean trypsin inhibitor type I-S has a molecular mass of 20.1 kDa suggests that the protease that lactoferrin affects is on the outside of the bacteria. It is very unlikely that such a large protein passed through the outer membrane
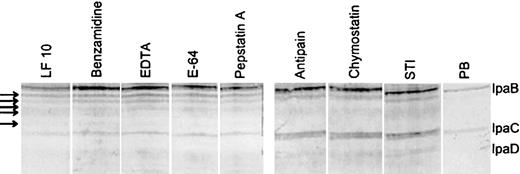
Effect of protease inhibitor treatment on lactoferrin-induced degradation of invasion antigens of Shigella flexneri Bacteria were incubated for 1 h in the presence of lactoferrin (0.125 mM) and protease inhibitors. Arrows indicate degradation products of IpaB induced by lactoferrin. The generation of these products was unaffected by protease inhibitors including benzamidine, EDTA, E64, or pepstatin in the incubation mix with bacteria and lactoferrin. In contrast, antipain, chymostatin, and soybean trypsin inhibitor blocked breakdown of IpaC and at least partly blocked degradation of IpaB. All experiments used identical nos. of bacteria
Lactoferrin does not cause degradation of previously released IpaB or IpaC To determine whether lactoferrin directly or indirectly causes degradation of IpaBC, an invasion plasmid antigen preparation that had been filter sterilized to assure that no bacteria were present was incubated with various concentrations of lactoferrin. IpaB and IpaC were not directly degraded by lactoferrin (figure 7). Because lactoferrin appears to have no proteolytic activity, it causes degradation of invasion proteins indirectly through its interaction with bacteria. The findings suggest that lactoferrin acts at the surface of the bacteria to cause IpaB and IpaC to become susceptible to surface protease(s)
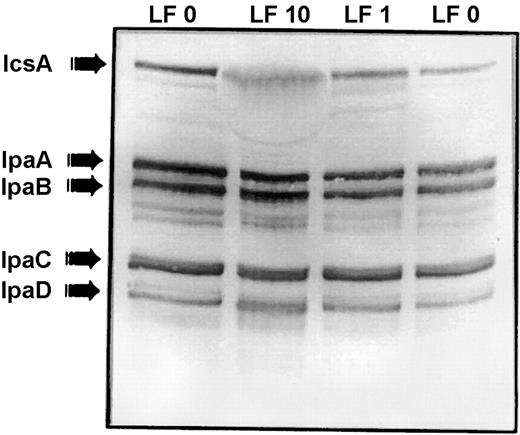
Lactoferrin does not cause degradation of previously released IpaB and IpaC. A cell-free invasion plasmid antigen water extract preparation was incubated for 1 h at 37°C with various concentrations of lactoferrin (LF) in PBS: PBS without lactoferrin (both LF 0 lanes) and with 10 mg/mL lactoferrin (lane LF 10) and 1 mg/mL lactoferrin (lane LF 1) This blot with preabsorbed convalescent serum that reacted with all of the invasion antigens showed that neither IpaB nor IpaC was directly degraded by lactoferrin. In contrast to studies in which lactoferrin treatment of bacteria led to degradation of IpaBC, lactoferrin without bacteria present did not degrade these invasins
Discussion
Virulence in Shigella species is encoded by a set of genes located primarily on a 230-kb plasmid [33, 34]. Within this large plasmid, a 31-kb region encodes for the invasion plasmid antigen genes (ipa) genes for mem-brane expression (mxi) [35], and surface presentation (spa) of invasion plasmid antigens [36, 37]. The ipa genes encode polypeptides (IpaA, IpaB, IpaC, and IpaD) that are the immunodominant antigens in the humoral response to Shigella infection [38–40]. Contact with epithelial cells activates this type III secretory system of Shigella species, initiating the secretion of invasion plasmid antigens that, in turn, cause bacteria to be taken up by the epithelial cells [29, 41]. This bacteria-directed phagocytic process involves participation of clathrin-coated pits [42]. After bacterial uptake, actin-mediated intracellular spread and infection of adjacent cells via membrane protrusions are encoded by IcsA, a surface-expressed ATPase [43–47]. Shigella mutants that do not produce IcsA have impaired virulence both in vitro in plaque-forming tissue culture invasion assays and in vivo in monkeys [48]
The critical first steps in bacteria-directed internalization in Shigella species are under the control of IpaB and IpaC [30]. These 2 invasins are transported to and expressed on the bacterial cell surface [31, 49] where the Mxi-Spa type III secretory mechanism secretes them. Both IpaB and IpaC are necessary for internalization of Shigella species [50]. IpaB and IpaC are unstable unless complexed intracellularly with IpgC, a molecular chaperone [31], or extracellularly with each other. IpaBC produced in response to contact with a eukaryotic cell is secreted as a stable complex. This complex alone is adequate to trigger uptake, since HeLa cells rapidly take up latex particles coated with IpaBC and induce actin polymerization that is dependent on small-ρ GTPases [30]. After secretion, they form pores in host cell membranes and remain closely associated, perhaps still complexed with each other [51]. Since IpaB and IpaC appear not to be synthesized during intracellular multiplication [52], lactoferrin-mediated loss of IpaB and IpaC prior to host cell contact could impair invasion and abort infection
Decreased invasiveness after lactoferrin exposure could be due to previously defined activities of lactoferrin. It binds bacterial porins [20, 53, 54], including those of Shigella species, and its antibacterial activity could be due in part to this binding. However, porins are not major virulence factors in Shigella species. Lactoferrin also acts on the cell surface to damage the outer membrane [55, 56]. Previous studies that used very sensitive radiolabeling methods demonstrated LPS release from bacteria after lactoferrin treatment [57]
Lactoferrin binds to LPS via 2 sites: a high-affinity N-terminal domain and a low-affinity site in the C-terminus [58]. The bactericidal activity of the pepsin-cleaved product is defined as a segment near the N-terminus consisting of an 18-aa loop formed by disulfide bonds between residues 20 and 37 [59]. Synthetic peptides corresponding to this cationic loop are rapidly bactericidal [60, 61]. Lactoferrin binds to the phosphate groups of lipid A, which makes the acyl chains more rigid so that they are more tightly packed [62]. N-terminal residues are essential for interaction with lipid A [63]. Within the N-terminus, a cationic cradle formed in the first 33 aa represents the minimal structure needed to bind lipid A and neutralize the effects of endotoxin [64]. Since this is the smallest defined lipid A binding sequence of lactoferrin, we focused on this portion of the molecule and on the whole protein. Because of the change in LPS packing induced by this cationic region, lactoferrin could disrupt interactions between LPS and cell surface proteins. LPS could have a role in organizing, orienting, and presenting the needle complex and in the secretion mechanism for the major virulence proteins
The loss of IpaB and IpaC in an unstable form after exposure to lactoferrin is unique because increased secretion of virulence proteins is linked to increased breakdown. Lactoferrin-induced degradation of the IpaBC requires that the complex be on the cell surface when it encounters lactoferrin. It was once thought that the IpaBC complex was formed in the supernatant of Shigella species cultures [65]. Recent observations suggest that IpaB and IpaC must be assembled at the cell surface before injection through the secretion needle [51]. The mxi/spa genes bring IpaB and IpaC together when a signal for their association and secretion is received. Our data suggest that lactoferrin disrupts the cell surface and exposes IpaB and IpaC to outer membrane proteases
BLAST searches reveal that lactoferrin and lactoferrin-binding proteins have some homology to type III secretory system proteins [66]. Lactoferrin may interact with either surface LPS or a type III secretory system protein and thereby interfere with the normal secretion mechanism and allow the invasins to be exposed to surface protease(s). It is unclear whether the relevant protease is IcsP (SopA), which is evenly distributed over the bacterial surface [67, 68], or some other protease. However, the fact that we could not demonstrate that IcsA was degraded in the presence of lactoferrin suggests that IcsP (SopA) was not involved since this protease is known to cleave IcsA. It has been suggested that lactoferrin has proteolytic activity that causes degradation of important colonization proteins (IgA protease and Hap) of Hemophilus influenzae [69, 70]. However, the experiments that suggested protease activity used intact live H. influenzae rather than isolated bacterial proteins. If we had not evaluated the effect of lactoferrin on the secreted virulence antigens, we also might have concluded that lactoferrin has proteolytic activity. Since lactoferrin lacks sequences usually found in proteases, a more likely explanation for the data is that lactoferrin is indirectly responsible for degradation of surface-expressed virulence proteins
The lipid A–binding N-terminal 33 aa peptide did not mimic the whole lactoferrin molecule in triggering degradation of invasion plasmid antigens. The fact that the lipid A binding the N-terminal portion of lactoferrin causes no breakdown of invasion plasmid antigens suggests a 2-step process triggered by lactoferrin. The first step is induction of secretion of invasion antigens by the N-terminal portion of the molecule; a second step involves some other portion of lactoferrin directly or indirectly causing a surface-expressed protease to cleave the invasion antigens
The virulence machinery of S. flexneri shares common elements with other enteropathogens. Enteroinvasive Escherichia coli possess virulence genes virtually identical to those of Shigella species[71]. The relationship between Shigella and Salmonella species virulence genes also is striking [72]. Salmonella typhimurium, Salmonella typhi and Salmonella dublin have chromosomally encoded invasion proteins homologous to IpaA–D of Shigella species, as well as genes closely related to the surface expression/secretion mxi/spa translocon of the type III protein secretion system [73–75]. Functionally, these invasion proteins are closely related: the Salmonella homologue of IpaB can restore invasiveness to a noninvasive Shigella IpaB mutant [76]. Among other Enterobacteriaceae, the Shigella secretion/translocation mechanism for invasion proteins is similar to that of Yersinia species [72, 77, 78] and enteropathogenic E. coli [79]. Surface expression and secretion of virulence proteins is the usual strategy shared by these bacterial enteropathogens. Thus, it will be important to determine whether the mechanism by which lactoferrin impairs virulence in S. flexneri is mimicked by similar effects on other gram-negative en-ter-ic pathogens that express closely related outer membrane–anchored virulence proteins
Acknowledgments
We acknowledge the technical assistance of Irene Herrera-Insua, Enrique Caceres, and Mustafa Siddiqui and thank E. V. Oaks for providing monoclonal antibodies 1H4, 2G2, and 16F8 and polyclonal rabbit antiserum to IcsA
References
Financial support: National Institutes of Health (grant HD-13021); Agennix.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}