
Abstract
Three sibling species of rough periwinkles are currently recognized: Littorina arcana, L. compressa and L. saxatilis. Certain forms of L. saxatilis are also argued by some to deserve species status, such as the barnacle-dwelling ‘L. neglecta’ and the lagoonal ‘L. tenebrosa’. Relationships between these taxa, and between and within representative populations, are investigated using sequence analysis and restriction fragment length polymorphism of a mitochondrial DNA fragment spanning the cytochrome oxidase I and cytochrome oxidase II gene boundary. These data show that there is some sharing of haplotypes between species, with L. arcana haplotypes paraphyletic with respect to L. saxatilis haplotypes, and L. compressa haplotypes paraphyletic to both L. arcana and L. saxatilis haplotypes. Such sharing of mtDNA haplotypes could be a consequence of either persistent hybridization or episodes of hybridization, or incomplete lineage sorting of ancestral polymorphisms. On the balance of evidence it is suggested that the latter, rather than hybridization events, is the more likely causal agent of the observed distribution. Intraspecific variation is extensive and it is suggested that the patterns of intraspecific polymorphism are explainable by a combination of historical factors (the impact of the Pleistocene ice-age) and contemporary restrictions to gene flow. It is argued that Littorina haplotypes evolved in at least two separate glacial refugia and became scattered by the subsequent range expansion around most of the coastline. Recent factors such as restricted gene flow and bottlenecks would then be capable of affecting the haplotype distribution, resulting in the pattern observed.
Similar content being viewed by others

Introduction
Periwinkles of the genus Littorina are familiar components of rocky shore communities. Around northern European coastlines six species are recognized. L. littorea (L.), the edible periwinkle, is a distant relative to the other five species, probably sharing an ancestor 12–15 Ma (Reid et al., 1996). These five are all members of the subgenus Neritrema, including two flat periwinkles L. fabalis (W. Turton) and L. obtusata (L.), and three sibling species of rough periwinkles, namely L. saxatilis (Olivi), L. arcana Hannaford Ellis and L. compressa Jeffreys. These five species have existed for a maximum of 3.5–4 Myr when their ancestral Littorina crossed from the Pacific into the Atlantic following the opening of the Bering Strait.
The age of the rough periwinkles is difficult to estimate, but reviewing allozyme evidence Reid et al. (1996) suggest that a time period of approximately 1.73 Myr (SE=0–3.55) is likely for this group.
The three rough periwinkle species show considerable allozyme polymorphism, which is apparently greatest in L. saxatilis (Ward, 1990). They lack a planktonic dispersal phase, and therefore show greater interpopulation genetic heterogeneity than do related snails with such dispersal (Janson, 1987). Further, their distribution on coasts differs: L. arcana is absent from the eastern two thirds of the south coast of England and L. compressa is particularly patchy in distribution; in contrast to these two, L. saxatilis is relatively ubiquitous on rocky coasts (Mill & Grahame, 1992). These distributions, together with the levels of inherent variation, population size and history, and low dispersal and gene flow, should have an influence on the genetic structure of the populations.
Allozyme electrophoresis studies of these species have failed to detect diagnostic loci, although there are frequency differences. Phylogenies based on allele frequencies are usually in agreement with morphological data and analysis of reproductive mode in suggesting that L. compressa is the least derived member of the group (Ward, 1990). However, from allozyme evidence Backeljau & Warmoes (1992) concluded that L. arcana was basal. Whichever is correct, it is certain that the species are closely related, and this, coupled with the existence of contentious forms, has led to the term L. saxatilis species complex as a descriptor of the group as a whole. Their close relationships are corroborated by the ability of male L. saxatilis and female L. arcana to hybridize under laboratory conditions (Warwick et al., 1990). Enzyme studies have been applied to the analysis of some of the contentious forms such as the barnacle-dwelling L. neglecta (Johannesson & Johannesson, 1990) and the lagoonal L. tenebrosa (Janson & Ward, 1985), with the conclusion that there is no evidence for species status, since neither diagnostic loci nor frequency shifts were evident. However, allozyme studies can be influenced by selection on the loci involved, moreover allozyme-inferred loci may require extensive time to reach a point at which separate gene pools are detectable, leading to low resolution. DNA based studies are potentially more powerful (Mitton, 1994), but few such studies have been undertaken on Littorina. Reid et al. (1996) established a mtDNA (cytochrome b, 12SrRNA and 16SrRNA) phylogeny of all extant Littorina but the resolution of the three rough periwinkle species was low, and there was no indication as to how inclusion of multiple individuals (and therefore potential intraspecific variation) affected the phylogeny. Kyle & Boulding (1998) reported on the use of mtDNA cytochrome b variation to examine population structure of L. subrotundata but, as yet, there have been no attempts to use mtDNA to examine population level variability in North Atlantic Littorina species. Evidence from maternally inherited mtDNA may allow analysis of introgression or of directionality of possible hybridization between closely related species, especially where comparable nuclear data sets are available.
Here we investigate the mitochondrial cytochrome oxidase I gene with respect to phylogeny, phylogeography, evidence for monophyly in the three currently recognized species, and the possibility of hybridization between them in the wild. We also ask what evidence these data provide concerning the taxonomic status of some contentious forms of L. saxatilis such as L. neglecta (e.g. Caley et al., 1995) and L. tenebrosa (e.g. Barnes, 1993).
Materials and methods
Sample collection
Samples of Littorina were collected from locations around the United Kingdom and Eire over the period between July 1996 and August 1998 (Table 1). Animals were collected from shores where locally abundant. Samples were taken over an area of at least 2 m2 in an effort to avoid sampling of single families. A single specimen of L. sitkana from Akkeshi, Hokkaido, Japan was utilized as an outgroup.
DNA extraction and PCR
Total genomic DNA was extracted from head-foot tissue of individual animals using protocol 47 of Ashburner (1989). DNA sample concentrations were measured by fluorometry and adjusted to 10 ng μL–1. PCR was performed in 50 μL volumes containing 50 mM KCl, 10 mM Tris-HCl pH 9.0, 1.5 mM MgCl2, 0.1% Triton X-100, 0.01% gelatin, 200 μM each dNTP, 25 pmol each primer, 25 ng DNA and 1 U Taq (Supertaq, HT Biotechnologies). The primer pair saxcoI and saxcoII, designed from sequence in Wilding et al. (1999) (accession number AJ132137), was used. SaxcoI covers bases 219–240 and saxcoII bases 1185–1166 of Wilding et al. (1999), and amplifies 967 bp (inclusive of primers) covering the 3′ end of cytochrome oxidase I, 30 bp of apparent noncoding sequence and the first 38 bp of CoII. PCR conditions of 1× 94°C, 5 min; 35× (94°C, 1 min, 55°C, 30 s, 72°C, 1 min); 1× 72°C, 5 min were employed. Positive PCR reactions were cleaned by PEG precipitation (Ausubel et al., 1990) and resuspended in 20 μL. Subsequently 4 μL was used in an automated sequencing reaction with the amplification primer saxcoII, using Taq terminator sequencing mix, and run on an ABI377.
Sequence analysis
Haplotype sequences were aligned in CLUSTAL W (Thompson et al., 1994). Identical haplotypes were identified through the construction of a distance matrix in MEGA v. 1.02 (Kumar et al., 1993) and removed from the dataset. Sequence variation was examined with DnaSP v. 2.2 (Rozas & Rozas, 1997). Phylogenies were constructed using both distance and maximum likelihood (ML) methodologies with L. sitkana as an outgroup. Haplotype relationships were inferred by both neighbour joining of Jukes–Cantor distances with 1000 bootstrapped data sets, implemented in MEGA v. 1.02 (Kumar et al., 1993) and by ML analysis performed by quartet puzzling (Strimmer & von Haeseler, 1996) using PUZZLE with 1000 quartet puzzling steps (analysis performed at http://bioweb.pasteur.fr/seqanal/interfaces/Puzzle.html). A haplotype network was also manually constructed detailing the relationships of haplotypes and the number of mutational steps required to interconvert haplotypes.
RFLP analysis
From aligned sequences, polymorphic restriction enzymes were chosen for which the recognition sequence spanned a variable site. Three suitable enzymes (DdeI, DraI and HindIII) were identified and employed to screen for variation. Digestions were performed in 15 μL volumes containing 5 μL PCR product, 1.5 mL 10× reaction buffer and 1–4 units enzyme at 37°C. Digestion products were separated on 2% agarose gels.
Estimates of θ (FST analogue) were calculated using FSTAT (Goudet, 1995) with standard errors calculated using jackknifing procedures. AMOVA and FST were performed in ARLEQUIN v. 1.1 (Schneider et al., 1997). For the AMOVA analysis, six groups were identified on the basis of regional differences in shell morphology and on distributional breaks: south-east, south-west, South Wales, Irish Sea, Ireland and East coast. Contingency tests were implemented in GENEPOP (Raymond & Rousset, 1995).
Results
Sequence analysis
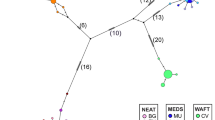
PCR using the primer pair saxcoI–saxcoII generated a 967-bp fragment. The single primer saxcoII was then used to sequence one end of this fragment. Four hundred base pairs of unambiguous sequence were generated from each of 95 separate individual Littorina representing a range of recognized species including 89 rough periwinkles (L. arcana, L. compressa and L. saxatilis and including some representatives of the contentious taxa L. neglecta, L. tenebrosa and L. saxatilis ‘b’), two L. fabalis, three L. obtusata and one specimen of Littorina sitkana (Table 2). Twenty-four distinct haplotypes were identified from these 95 individuals (Table 3). Sequences are deposited in Genbank with accession numbers AJ133311–133334. Of the 400 bp of sequence, 56 sites exhibited variation, all involving individual substitutions with the exception of a single base indel difference between the L. sitkana sequence and all other sequences. When L. sitkana is removed there are 37 variable sites of which 25 are parsimony informative. If only the rough periwinkles are considered these figures drop to 28 variable sites with 16 parsimony informative positions. All substitutions are silent with the exception of the G → A transition at position 938 which alters the amino acid from valine to isoleucine, and the T → G transversion at position 1070 which changes the amino acid from serine to alanine. Because of the low number of parsimony informative sites, parsimony-based phylogeny reconstruction methods were considered inappropriate and haplotype phylogenies were constructed using both a distance method and a maximum likelihood variant. The resultant trees (Fig. 1A and B) are essentially congruent, although the distance-based tree suggests some details of branching order that are not seen in the maximum likelihood tree. Bootstrap support values are generally low, although there are some well supported nodes.

(A) Neighbour-Joining tree of Jukes–Cantor distances for the 24 CoI haplotypes of Littorina. Numbers at nodes refer to percentage bootstrap support from 1000 replicates. (B). Quartet-puzzling maximum likelihood tree constructed using the HKY model in PUZZLE. Numbers at nodes indicate how many times the cluster to the right of the node was found in 1000 quartet puzzling steps.
Disregarding the outgroup (L. sitkana, haplotype 24) and the sequences from flat periwinkles (haplotypes 22 and 23), there are four main groups discernible. The haplotypes limited to L. compressa (17–21) form a single group, and haplotypes 15 and 16 which are mainly limited to L. arcana (and some east coast L. compressa) also group together. The remaining L. saxatilis sequences (in a few instances these also occur in L. arcana or L. compressa — see later for details) form two main groups. One group (termed L. saxatilis group β) is well supported in both phylogenies (haplotypes 10–14). The remaining sequences (1–9) form a single group in the distance method, whilst in the ML tree they are unresolved, with the exception of a group of three (7–9). Haplotypes 1–9 are labelled L. saxatilis group α. The lack of resolution in the phylogeny of haplotypes of group α is a consequence of these sequences differing from each other by only singleton base changes. Although the support values within the L. saxatilis haplotypes are low, the two distinct groups of L. saxatilis haplotypes are also revealed in the haplotype network (Fig. 2). Here the four separate haplotype groups (the ‘compressa’ group, the ‘arcana’ group and L. saxatilis groups α and β) distinguishable in the phylogenies can be discerned (surrounded by the dashed lines). This haplotype network is not a minimum spanning tree in that individual haplotypes are not separated by single substitutions, and there are many ambiguous connections. This is probably a consequence of not having uncovered all existent haplotypes, and perhaps also due to homoplasies in the data (Besansky, 1997; Brown Gladden et al., 1997), although such events seem less likely when haplotypes of closely related species are being compared.

Haplotype network showing the number of base changes between 24 CoI haplotypes of Littorina. Bars on interconnecting lines represent the number of mutational steps. Dashed lines encircle major groups (see text).
Most haplotypes are unique to a particular species, with the exceptions of haplotypes 1 and 10, which are seen in L. saxatilis, L. arcana and L. compressa, and haplotype 15, which has been encountered in both L. arcana and L. compressa. Representative L. neglecta, L. tenebrosa and L. saxatilis ‘b’ did not have distinct haplotypes, but shared haplotypes with L. saxatilis (Table 2).
Restriction digestion
From sequence information three informative restriction enzymes were identified, DdeI, DraI and HindIII. These released 1–3 fragments per enzyme (Table 4). Individual haplotypes were designated by single letter codes and a three-letter composite haplotype was then generated for each animal. There were insufficient informative enzymes to differentiate every sequenced haplotype. However, the three enzymes used allowed resolution into the main groups obvious from the haplotype network and phylogenies. Six separate composite haplotypes were uncovered. The L. compressa group (17–21) are all BAB and the L. arcana haplotype group (15–16) all BAA. For L. saxatilis group α, haplotypes 2 and 5 are BCA and the remainder are BBA, whilst for group β, haplotypes 10 and 12–14 are AAA and haplotype 11 is ADA. A stylised tree based on Fig. 1A and B and summarizing the composite haplotype information is depicted in Fig. 3. Table 5 and Fig. 4 together describe the distribution of composite haplotypes within populations of the various species. Because sample sizes are much higher than those utilized for the sequencing study, this RFLP analysis gives a much better picture of the geographical distribution of haplotypes. In L. arcana there is a definite east–west split, with all east coast members having the BAA haplotype and the majority of west coast animals being AAA. Thus there is a geographical pattern to those haplotypes that are shared with L. saxatilis. East coast animals (St Abb’s, North Berwick and Old Peak) all have the ‘L. arcana’ haplotype BAA, whilst those on the west coast often have haplotypes typical of L. saxatilis, such as AAA. For L. compressa the picture is less clear and variation is higher than in L. arcana. In this species there seems to be no obvious pattern to those haplotypes that are shared with either L. arcana or L. saxatilis. L. compressa from the Lizard, Trevaunance and Inismór are fixed for the ‘compressa’ haplotype, but elsewhere animals with ‘saxatilis’ haplotypes (BCA, BBA, AAA) are common. Because of more intensive sampling, there is more information on the geographical variation of haplotypes in L. saxatilis. Two things are apparent. Firstly, as for L. arcana there is a difference between east coast and west coast animals, with the BBA haplotype much more frequent on the west coast, and a higher frequency of BCA and AAA in the east. Secondly, there is a south-west enclave of L. saxatilis with a high frequency of the ADA composite haplotype. This haplotype is seen nowhere else in the country. F-statistics (θ and FST) indicate substantial population subdivision (Table 6) and this is corroborated by contingency tests which show significant differences among populations of all three species (P=0.000 ± 0.000). AMOVA analysis (Table 6) using the preordained groups identified on the basis of shell morphology and known disjunctions in littorinid distribution, indicates varying degrees of evidence for mtDNA variability being partitioned among these groups. Although for L. arcana over 70% of the total variation is among these groups, the figure is somewhat misleading, since virtually all variation is partitioned solely on an east coast–west coast basis, and the choice of groups reflects this to an extent. For L. compressa and L. saxatilis, substantial variation is within populations, reflecting high levels of polymorphism. For L. saxatilis, little of the remainder of the variation is among groups, indicating that these do not explain the variation encountered. In L. compressa nearly 40% of the variation is among groups, but as for L. arcana, this is probably attributable to east–west division, whereby most west coast L. compressa show the ‘compressa haplotype’ whereas a higher proportion of east coast animals display the ‘saxatilis’ or ‘arcana’ type.

Stylized dendrogram depicting relationships of RFLP composite haplotypes of Littorina.

Geographical variation in CoI RFLP patterns for Littorina saxatilis, L. arcana and L. compressa. Areas of pie charts represent composite haplotype frequency.
Discussion
The phylogeny of Littorina mitochondrial CoI haplotypes
The deduced branching order of the Littorina species considered here is in agreement with current, consensus opinion (Reid, 1996). The low bootstrap support in the phylogeny at nodes leading to the three rough periwinkle species could be a consequence of a period of rapid speciation, with only short time periods between speciation events during which informative substitutions could accrue. Periods of rapid speciation have similarly affected phylogeny construction in finches (Fehrer, 1996) and corvids (Helm-Bychowski & Cracraft, 1993). Paraphyletic haplotype distributions are also apparent with L. obtusata haplotypes paraphyletic with respect to L. fabalis, L. arcana haplotypes paraphyletic with respect to L. saxatilis, and L. compressa haplotypes paraphyletic with respect to both L. arcana and L. saxatilis haplotypes. Such paraphyly may have arisen as a consequence of shared ancestral polymorphisms when (usually moderate to large) populations have not progressed to reciprocal monophyly during speciation. However introgressive hybridization may also be involved (Besansky, 1997; Schneider-Broussard et al., 1998) and it is well documented that mtDNA can cross species boundaries through hybridization or introgression (Avise, 1994). If hybridization were the causal mechanism of the observed haplotype distribution, this would require viability of hybridization between all three species. Although Warwick et al. (1990) showed laboratory hybridization in one direction (female L. arcana with male L. saxatilis), there is no information on the mating preferences during back-crosses, and there were no successful hybridizations with other species crosses. We suggest that this asymmetrical haplotype distribution, with ‘L. saxatilis’ haplotypes observed in L. arcana and L. compressa but no ‘L. compressa’ haplotypes or ‘L. arcana’ haplotypes found in L. saxatilis, rules out general hybridization (i.e. occasional gene flow between the species). Although introgression of the mtDNA of one species into another is not precluded, it seems unlikely on the basis of existing evidence.
It is also the case that the animals with haplotypes typical of another species have a restricted distribution. For instance, L. arcana with the ‘saxatilis’ haplotype occur on the west coast of Britain, but have not been found on the east coast (Fig. 4, Table 5), and L. compressa with particular ‘arcana’ or ‘saxatilis’ haplotypes are patchy. In most cases these haplotypes are not the commonest ones, and in some cases are not present sympatrically in the other species, e.g. all West Dale Bay L. arcana have a ‘saxatilis’ haplotype AAA, but all West Dale Bay L. saxatilis are BBA. This again suggests that localized hybridization is not the cause of the observed haplotype sharing, unless the hybridization happened in the past when, perhaps, haplotype frequencies were very different.
The alternative explanation, retention of ancestral polymorphisms, has been documented for mtDNA genes in other groups (e.g. Helm-Bychowski & Cracraft, 1993; Fehrer, 1996; Schneider-Broussard et al., 1998), in which shared haplotypes antedate speciation and are haphazardly maintained in certain populations through incomplete lineage sorting. Rapid and recent speciation (see above) is consistent with this since there will have been little time for sorting of ancestral haplotypes into the descendant taxa. This hypothesis also fits with the ‘directional’ nature of haplotype sharing. The least derived littorinid, L. compressa, displays haplotypes representative of all groups currently recognized as ‘L. compressa type’, ‘L. arcana type’ and ‘L. saxatilis type’. A more limited range of haplotypes were passed on to the descendant L. arcana and L. saxatilis, being enriched with respect to what has become the ‘arcana type’ and the ‘saxatilis type’ whilst still leaving historical signatures — ‘arcana type’ and ‘saxatilis type’ haplotypes in L. compressa and ‘saxatilis type’ haplotypes in L. arcana. The regional distribution of shared haplotypes is also consistent with this mechanism. Since all three species are either ovoviviparous or oviparous, and therefore undergo reduced gene flow, they will be more prone to bottlenecks and genetic drift, which would, over the time since speciation, have let some populations approach fixation for species-characteristic haplotypes ahead of others.
Watterson & Guess (1977) have argued that the most frequent alleles (haplotypes) may be the oldest; they will consequently be most likely to be shared. It is the case here that the shared haplotypes (i.e. numbers 1, 10 and 15; Table 3) are three of the six most frequent.
Lineage sorting should be quicker for mtDNA than for nuclear DNA and thus, if there is incomplete sorting of mtDNA haplotypes resulting in paraphyly, then this should be particularly evident in the case of nuclear alleles (Besansky, 1997). From data on nuclear DNA RFLPs at two calmodulin introns and two anonymous loci (Wilding et al., unpubl. data) there is evidence that alleles at nuclear loci are incompletely sorted. They are not homogeneous between species, which again suggests that unconstrained gene flow between species is not occurring (Clarke et al., 1996); neither are they fully diagnostic, suggesting mtDNA has not introgressed from a fully diagnostic nuclear background. In view of all these considerations, we suggest that the observed paraphyly is most likely due to incomplete lineage sorting.
Phylogeography of cytochrome oxidase I haplotypes
Population-level differences in the frequencies of composite haplotypes are evident within all three currently recognized species. Much of the variation is within populations, particularly for L. saxatilis and L. compressa, but there are significant differences between populations of all three species. Although there appear to be regional differences, the exact nature of these, and the factors responsible are difficult to disentangle, although an east coast — west coast differentiation seems apparent, particularly so for L. saxatilis and L. arcana. The distribution of these haplotypes is particularly complex for the more intensively sampled L. saxatilis. From the haplotype network and phylogenies, two groups of L. saxatilis haplotypes are recognizable. Similar haplotype networks occur in anchovy (Magoulas et al., 1996) and beluga whale (Brown Gladden et al., 1997), and have been argued to result from the origination of haplotypes in different parts of the species range. However, haplotypes from both clusters in L. saxatilis can be found around the sampled range, and the current distribution of L. saxatilis in the range studied is largely continuous, with no obvious disjunctions on either side of which evolution could occur in relative isolation. However, historically, this may not have been so, particularly during ice ages. The concept of glacial refugia in which species could survive (Taberlet et al., 1998) seems less appropriate to intertidal species, since it seems likely that ice sheets and permafrost should affect land biota more severely than species in the more buffered marine environment. However, it is without question that the distribution of Littorina spp. during the recent ice ages must have been considerably different from their current distribution, since sea levels were considerably lower, and glaciers extended from Scandinavia to Britain and through the Irish Sea (Bowen et al., 1985). Indeed, virtually all of the present range of Littorina considered in this study was unavailable during the last glacial maximum; thus their distribution must have been radically different. The presence of two groups of L. saxatilis haplotypes (α and β) may therefore be explainable by contemporary L. saxatilis haplotypes having evolved in two distinct areas, from which animals have subsequently dispersed and intermingled. Indeed postglaciation scattering of haplotypes has been argued to explain the observation that mtDNA haplotypes in the song sparrow Melospiza melodia were geographically widespread, such that closely related haplotypes were not geographically proximal (Zink & Dittmann, 1993). Similarly, range expansion from different refugia would explain why Littorina haplotypes from the two divergent groupings are found virtually across the species range. The situation in L. compressa and L. arcana is complicated by the presence of shared haplotypes, but, given the similarity in biology and ecology with L. saxatilis, similar factors, both historical and current will have moulded the pattern of haplotype variation in these species. Uncovering further evidence for this refugium hypothesis requires additional sampling of L. arcana, L. compressa and L. saxatilis, particularly from around mainland Europe (the site of potential refugia) to unravel the effects of historical and other factors on the distributions of mtDNA variation in these, and related, species.
Although the observed distribution of haplotypes in these species undoubtedly has some historical basis, because there is no planktonic dispersal phase the population genetic structure of these animals will also be profoundly influenced by low gene flow and small population sizes. A probable example of the effects of the levels of gene flow is seen for the ADA haplotype (haplotype 11 in the sequence study) of L. saxatilis, which has a very restricted distribution (in this case to south-western Britain). This seems to be a haplotype that has arisen via mutation on the south coast, remaining restricted to this area due to low gene flow. Genetic drift and inbreeding may also be important considerations due to the life histories of these species, and this may explain the wide variance in haplotype frequencies seen even in neighbouring populations (Fig. 4).
The status of L. neglecta, L. saxatilis ‘b’ and L. tenebrosa
CoI data provide no evidence to suggest that L. neglecta and L. saxatilis ‘b’ are anything other than morphological or ecological variants of L. saxatilis. No diagnostic haplotypes are seen. However, there are some differences in haplotype frequencies between L. tenebrosa and neighbouring populations of L. saxatilis.
References
Ashburner, M. (1989). Drosophila: A Laboratory Manual, Cold Spring Harbor Laboratory Press.
Ausubel, F., Brent, R., Kingston, R., Moore, D., Seidman, J., Smith, J. and Struhl, K. (1990). Current Protocols in Molecular Biology. Wiley, New York.
Avise, J. C. (1994). Molecular Markers, Natural History, and Evolution. Chapman & Hall, New York & London.
Backeljau, T. and Warmoes, T. (1992). The phylogenetic relationships of ten Atlantic littorinids assessed by allozyme electrophoresis. In: Grahame, J., Mill, P. J. and Reid, D. G. (eds) Proc. 3rd Int. Symp. Littorinid Biol, pp. 9–24. Malacological Society of London, London.
Barnes, R. S. K. (1993). On the nature of the coastal lagoon winkles attributed to Littorina tenebrosa and Littorina saxatilis. Cahiers Biol Mar, 34: 477–495.
Besansky, N. J. (1997). Patterns of mitochondrial DNA variation within and between African malaria vectors, Anopheles gambiae and An. arabiensis suggest extensive gene flow. Genetics, 147: 1817–1828.
Bowen, D. Q., Rose, J. and McCabe, A. M. (1985). Correlation of quaternary glaciations in England, Ireland, Scotland and Wales. Quart Sci Rev, 5: 299–340.
Brown Gladden, J. G., Ferguson, M. M. and Clayton, J. W. (1997). Matriarchal genetic population structure of North American beluga whales Delphiapterus leucas (Cetacea: Monodontidae). Mol Ecol, 6: 1033–1046.
Caley, K., Grahame, J. and Mill, P. J. (1995). A geographically-based study of shell shape in small rough periwinkles. Hydrobiologia, 309: 181–193.
Clarke, B., Johnson, M. S. and Murray, J. (1996). Clines in the genetic distance between two species of island land snails: how ‘molecular leakage’ can mislead us about speciation. Phil Trans R Soc B, 351: 773–784.
Fehrer, J. (1996). Conflicting character distribution within different data sets on cardueline finches: artifact or history? Mol Biol Evol, 13: 7–20.
Goudet, J. (1995). Fstat (version 1.2): a computer program to calculate F-statistics. J Hered, 86: 485–486.
Helm-Bychowski, K. and Cracraft, J. (1993). Recovering phylogenetic signal from DNA sequences: relationships within the corvine assemblage (Class Aves) as inferred from complete sequences of the mitochondrial DNA cytochrome-b gene. Mol Biol Evol, 10: 1196–1214.
Janson, K. (1987). Allozyme and shell variation in two marine snails (Littorina Prosobranchia) with different dispersal abilities. Biol J Linn Soc, 30: 245–256.
Janson, K. and Ward, R. D. (1985). The taxonomic status of Littorina tenebrosa Montagu as assessed by morphological and genetic analysis. J Conch, 32: 9–15.
Johannesson, K. and Johannesson, B. (1990). Genetic variation within Littorina saxatilis (Olivi) and Littorina neglecta Bean: is L neglecta a good species? Hydrobiologia, 193: 89–97.
Kumar, D., Tamura, K. and Nei, M. (1993). Mega: Molecular Evolutionary Genetic Analysis The Pennsylvania State University, University Park, PA.
Kyle, C. J. and Boulding, E. G. (1998). Molecular genetic evidence for parallel evolution in a marine gastropod, Littorina subrotundata. Proc R Soc B, 265: 303–308.
Magoulas, A., Tsimenides, N. and Zouros, E. (1996). Mitochondrial DNA phylogeny and the reconstruction of the population history of a species: the case of the European anchovy (Engraulis encrasicolus). Mol Biol Evol, 13: 178–190.
Mill, P. J. and Grahame, J. (1992). Distribution of the rough periwinkles in Great Britain. In: Grahame, J., Mill, P. J. and Reid, D. G. (eds) Proc. 3rd Int. Symp. Littorinid Biol, pp. 305–307. Malacological Society of London, London.
Mitton, J. B. (1994). Molecular approaches to population biology. Ann Rev Ecol Syst, 25: 45–69.
Raymond, M. and Rousset, F. (1995). Genepop (version 1.2): population genetics software for exact tests and ecumenicism. J Hered, 86: 248–249.
Reid, D. G. (1996). Systematics and Evolution of Littorina. The Ray Society, London.
Reid, D. G., Rumbak, E. and Thomas, R. H. (1996). DNA, morphology and fossils: phylogeny and evolutionary rates of the gastropod genus Littorina. Phil Trans R Soc B, 351: 877–895.
Rozas, J. and Rozas, R. (1997). DnaSP, version 2.0: a novel software package for extensive molecular population genetics analysis. CABIOS, 13: 307–311.
Schneider, S., Kueffer, J. M., Roessli, D. and Excoffier, L. (1997). Arlequin, version 1.1: a software for population genetic data analysis Genetics and Biometry Laboratory, University of Geneva, Switzerland.
Schneider-Broussard, R., Felder, D. L., Chlan, C. A. and Neigel, J. E. (1998). Tests of phylogeographic models with nuclear and mitochondrial DNA sequence variation in the stone crabs, Menippe adina and Menippe mercenaria. Evolution, 52: 1671–1678.
Strimmer, K. and von Haeseler, A. (1996). Quartet puzzling: a quartet maximum-likelihood method for reconstructing tree topologies. Mol Biol Evol, 17: 964–969.
Taberlet, P., Fumagalli, L., Wust-Saucy, A. -G. and Cosson, J. -F. (1998). Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol, 7: 453–464.
Thompson, J. D., Higgins, D. G. and Gibson, T. J. (1994). Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucl Acids Res, 22: 4673–4680.
Ward, R. D. (1990). Biochemical genetic variation in the genus Littorina (Prosobranchia: Mollusca). Hydrobiologia, 193: 53–69.
Warwick, T., Knight, A. J. and Ward, R. D. (1990). Hybridisation in the Littorina saxatilis species complex (Prosobranchia: Mollusca). Hydrobiologia, 193: 109–116.
Watterson, G. A. and Guess, H. A. (1977). Is the most frequent allele the oldest? Theor Pop Biol, 11: 141–160.
Wilding, C. S., Mill, P. J. and Grahame, J. (1999). Partial sequence of the mitochondrial genome of Littorina saxatilis: relevance to gastropod phylogenetics. J Mol Evol, 48: 348–359.
Zink, R. M. and Dittmann, D. L. (1993). Gene flow, refugia, and evolution of geographic variation in the song sparrow (Melospiza melodia). Evolution, 47: 717–729.
Acknowledgements
We wish to thank Dr David Reid of the Natural History Museum, London for providing the samples of L. sitkana and Cape Cornwall L. compressa, David Reid and Kerstin Johannesson for the Swedish L. saxatilis, and Dr Del Smith for provision of the Maine sample of L. saxatilis. This research was supported by the MAST-3 programme of the European Commission under contract MAS3-CT95–0042 (AMBIOS).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wilding, C., Grahame, J. & Mill, P. Mitochondrial DNA CoI haplotype variation in sibling species of rough periwinkles. Heredity 85, 62–74 (2000). https://doi.org/10.1046/j.1365-2540.2000.00718.x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1046/j.1365-2540.2000.00718.x
