




Abstract
Four specifically designed IDE mutants have been used to unveil the molecular basis of peptidase versus E1-like activity of the enzyme. We have found that physiological concentrations of copper(ii) ions inhibit the proteolytic activity of the enzyme towards small and large substrates but have no effect on the E1-like activity of the enzyme.
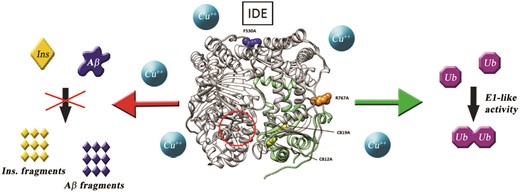
Four specifically designed IDE mutants have been used to unveil the molecular basis of peptidase versus E1-like activity of the enzyme.
Copper plays an important role in the biomolecular pathways responsible for the development of several pathologies such as Alzheimer’s disease. Insulin-degrading enzyme (IDE) is a metalloprotease capable of cleaving several different peptides and its activity as a proteolytic enzyme can be quenched by copper(ii). However, IDE is also able to activate ubiquitin molecules to form di-ubiquitin. Here, we have discovered that copper(ii) is able to divert enzyme activity towards the ubiquitin activation pathway and we have used IDE mutants to assess, at a molecular level, which portions of the enzyme are mainly responsible for the two different IDE functions.
Introduction
Insulin-Degrading Enzyme (IDE) is a zinc metalloprotease of the inverzincin family that has been first studied for its ability to degrade many amyloidogenic substrates such as insulin,1–3 amylin,4,5 amyloid beta peptides6 and many others.7,8 However, it was soon clear that IDE functions and activity go far beyond the simple role as a metalloprotease.9 Indeed, the enzyme has been demonstrated to work as a heat-shock protein,10 as an allosteric modulator of the 20S proteasome11 and as an activator of ubiquitin (Ub), promoting the formation of K48 and K63 diubiquitin.12 The ubiquitous presence of IDE in several tissues as well as in many different biological compartments, including endosomes, peroxisomes, mitochondria and the cell surface, confirms the multi-functional nature of this enzyme. Since IDE seems to be mainly located in the cytosol, in order to carry out its proteolytic function towards the above mentioned amyloidogenic substrates, the latter have to undergo internalization upon binding to their membrane receptor13 or IDE has to be secreted. However, the latter hypothesis is controversial and there is evidence that IDE is not secreted from cultured cells.14 Moreover, although the interaction of IDE with Ub has been widely reported15,16 and the Ub residues involved in such interaction have been unveiled,12 the structural features of IDE interactions with Ub and the biological significance of such interactions are not well understood.17
In this scenario, it is imperative to have insight into the molecular basis and into the enzyme structural requirements to carry out the different biological functions listed above. In other words, which, if any, molecular mechanisms are responsible for diverting the enzyme activity towards one function or the other? In an attempt to answer to this question, we have used four different IDE mutants, which have been chosen to give insight into specific molecular issues: R767A (exists mostly as a monomer), F530A (mutation in the linker region between IDE-N and IDE-C, promoting the open conformation of the enzyme), and C812A and C819A (mutations of the cysteine residues located near the C-terminal of IDE and close to the catalytic site).18 These mutants have been tested, together with the wild type (wt) form of the enzyme, for their capability to degrade long (Aβ1–40, amyloid beta peptide) and short (B20–30, a portion of the insulin B chain)19 peptides, as well as for their Ub activating (E1-like) activity.12 The two peptides, both degradable by IDE but having different chain lengths, have been used because of their different degradation kinetics observed in the case of short versus long peptides, attributed to the presence of an exosite on IDE, made of conserved residues and located ∼30 Å distant from the catalytic Zn2+.20 Indeed, while long substrates bind to the catalytic site as well as to the exosite, short substrates cannot bind both sites simultaneously and show peculiar kinetic features (higher Km values, i.e. lower affinity) and a generally higher rate of catalysis.20 However, it has been already reported that B20–30 is a substrate with a rather slower catalytic rate when compared with full length insulin and it has been chosen in this work mainly for its low affinity towards metal ions.19 By the way, copper and zinc have been widely reported to bind Aβ peptides, affecting their oligomerization and toxicity.21,22 They have also been reported to being able to affect IDE proteolytic activity in rather opposite ways,23 and their dyshomeostasis is now considered a prominent feature in many neurodegenerative diseases.24,25 In this work, we have also investigated how the proteolytic and the E1-like activity of IDE are affected by these metal ions.
The proteolytic activity of the IDE mutants
The results obtained by CD investigation carried out on the various IDE mutants are reported in Fig. S1 (ESI†). It is evident that, despite the single point mutations, the conformation of IDE is not significantly affected, as the enzyme maintains its structural features. However, the activities of the various mutants towards the B20–30 peptide are quite different from the activity of the wt, as reported in Table S1 (ESI†). Indeed, all the IDE mutants show a much lower activity than the wt enzyme (especially in the case of the R767A, C812A and C819A mutants), and very long incubation times (48 h) have been necessary to appreciably detect the B20–30 degradation pattern. For the same reason, although it would have been useful to obtain all the kinetic parameters of the B20–30 degradation for each IDE mutant, it was only possible to assess in a semi-quantitative way the overall activity for each IDE mutant in comparison with the wt and if there was a different response of the mutants to the two metal ions tested (the last column of Table S1, ESI†).
Analogously, the activity of IDE towards a much longer peptide, Aβ1–40, is also lower for all the mutants tested in comparison to the wt. As already reported, the IDE-mediated hydrolysis of the monomeric form of Aβ1–4026 produces many peptide fragments (Table S2, ESI†). Most of them form when IDE induces Aβ degradation at 19Phe-20Phe and 14His-15Gln cleavage sites. Minor cleavage sites involve the vicinal His residues and the C-terminal side of 16Lys, 18Val, 20Phe and 30Ala (Fig. 1). F530A is able to degrade Aβ, though to a lesser extent compared to the wt isoform. All the other IDE mutants tested (R767A, C812A, C819A) showed negligible or no effect on the amyloid peptide degradation (Table S2, ESI†). As reported in Fig. S2 (ESI†), the hydrolytic species of Aβ1–40 in the presence of F530A are equal to those formed by the action of wt IDE. The main difference between the wt form and the mutant relies on the relative intensities of the hydrolytic pattern. Aβ1–19 is the main species formed after 60 min reaction of the wt IDE-mediated hydrolysis, whereas F530A mainly induces the formation of Aβ1–14. F530A and wt IDE also differ in their activity upon the involvement of copper(ii) and zinc(ii) ions. Fig. S3 (top) (ESI†) shows that copper(ii) clearly inhibits the formation of all the peptide fragments, as previously reported.20 The extent of this overall decrease does not exceed 10–15% of the original value. On the other hand, zinc(ii) ions do not have any significant effect on the wt IDE-mediated hydrolysis of Aβ1–40. As for the activity of F530A (Fig. S3 (ESI†), bottom), copper(ii) still inhibits the Aβ1–40 degradation, whereas zinc(ii) only partially rescues the Aβ degrading activity of F530A compared to that shown by the wt form.
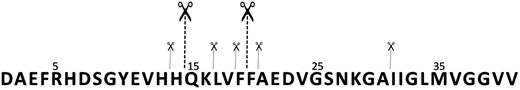
Depiction of the main (bigger scissors) and secondary (smaller scissors) cleavage sites of the IDE-mediated hydrolysis of Aβ1–40 at 37 °C after 1 h reaction.
The ability of the IDE mutants to activate ubiquitin
We have then tested the IDE mutants as ubiquitin activating enzymes, monitoring the pyrophosphate (PPi) release during the Ub activation process. Among all the scrutinized IDE mutants, only the C819A and C812A mutants show a clear decrement of PPi formation, whereas F530A and R767A have an increased activity in comparison to the wt as ubiquitin activating enzymes (Fig. 2).
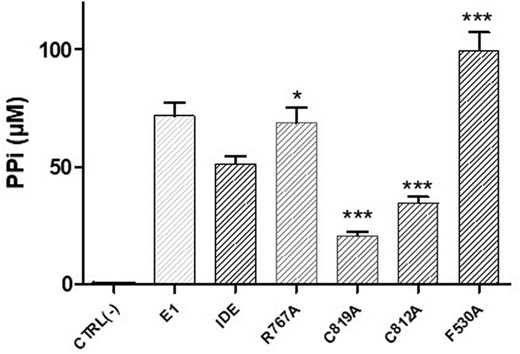
Lys48-linked polyubiquitination reactions were performed at pH 7.4 (T = 37 °C) in small volumes (40 μL) of ligation buffer (50 mM Tris–HCl, 5 mM MgCl2, 30 μM DTT and 2 mM ATP) containing Ub (10 μM), E2–25k (1 μM), IDE or IDE mutants (2 μM). The amounts of reaction mixtures (10 μL) were collected in a 96 well plate and analyzed using a Varioskan plate reader. In each well, 10 μL of a 400 mM EDTA solution were used to quench all reactions. The pyrophosphate concentration was determined with colorimetric assay.
As the C819A and C812A mutants show similar behaviours, we have then used only the former, together with the R767A and F530A mutants, to perform poly-ubiquitination reactions as indicated in the ESI.† From Fig. S4 (ESI†), it is possible to see that, as in the case of the wt enzyme,11 only the formation of Ub dimers is observed for the R767A and F530A mutants, while C819A does not function as an E1-like enzyme, revealing the direct involvement of cysteine 819 in the interaction with Ub.
The IDE-promoted Ub activation has also been studied in the presence of different concentrations of copper(ii) and zinc(ii) ions to establish how metals could affect the E1-like IDE activity.
In preliminary experiments, we have verified that copper(ii) and zinc(ii) do not interfere with PPi determination at the same concentration of metals used in these experiments (Fig. S5, ESI†). In Fig. 3, the PPi formation during Ub chain elongation reactions by the wt enzyme is reported and it is evident that the IDE E1-like activity is not affected by zinc(ii), whereas copper(ii) has an appreciable effect only at concentrations ≥20 μM.
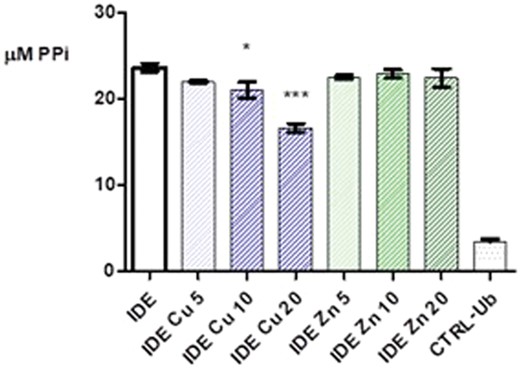
Pyrophosphate formation during Ub chain elongation reactions using IDE as a Ub activating enzyme, in the presence of different concentrations of copper(ii) (blue bars) and zinc(ii) (green bars): 5, 10 and 20 μM. Reported data are representative for three sets of independent experiments performed in triplicate.
The same experiments were performed using the IDE mutants and the obtained results are reported in Fig. S6 (ESI†). It is evident that the presence of metal ions does not have an appreciable effect on the E1-like activity of the investigated IDE mutants.
IDE structure–activity relationship
The effect that single residue mutagenesis has on the two main reported functions of IDE has been investigated via different experimental approaches. In Fig. 4 the structure of IDE in the closed state is depicted and the mutations taken into consideration in this work are highlighted. Although among the mutants considered in this work there were significant differences in the measured proteolytic activity, we have found that all the mutations lower the proteolytic activity of the wt IDE towards long as well as short substrates. This result indicates the direct involvement of several different regions of the enzyme performing the proteolytic action and is somewhat different from what has been previously reported.14 However, the discrepancy could be ascribed to the differences in the substrates used for the degradation assays. Indeed, while for substrates as short as B20–30 a significant activity is detected for the mutants considered, for longer substrates such as Aβ1–40, which are supposed to interact with the catalytic site as well as the exosite of the enzyme, some of the mutants resulted to be inactive. More importantly, the E1-like activity of IDE varies consistently depending on the single point mutation considered. Indeed, while F530A and R767A show an increased E1-like activity when compared to the wt form of the enzyme, the C812A and C819A mutants are mostly inactive. These results unambiguously demonstrate that the subdomain encompassing residues 781–981 of the enzyme is directly and mainly involved in (i) the E1-like activity of IDE and (ii) the proteolytic activity of the enzyme towards long substrates. The latter finding points out that, besides the recognized IDE exosite (residues 336–342 and 359–363),20 residues 781–981 also play an important role in the binding and processing of long substrates to IDE.
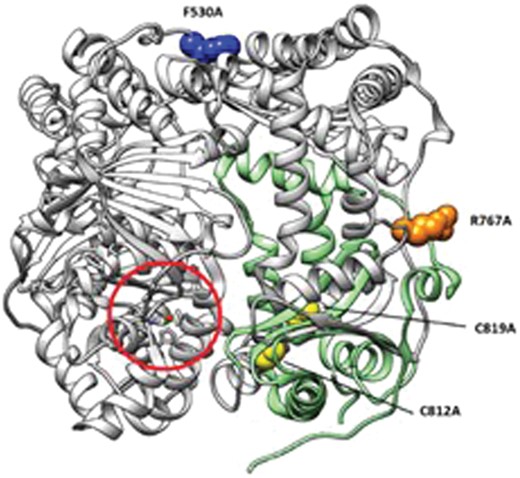
Structure of IDE (gray ribbons) in the closed state (PDB code: 2JG4). The zinc(ii) catalytic site is circled in red. Single residue mutations analyzed in this work are represented as blue (F530A), orange (R767A) and yellow (C812A and C819A) spheres. The subdomain encompassing the region which is mainly involved in E1-like activity (residues 781–981) is represented as green ribbons.
The effect of metal ions on the two investigated IDE functions
An important aspect investigated in this work was the effect that metal ions (copper and zinc) have on the two different IDE activities (proteolytic and E1-like) scrutinized. It is important to highlight that, under the experimental conditions used in this work, the observed effects are due to the direct binding of the metal ions to the enzyme and not to the formation of metal ion–peptide complexes.19 The results clearly show that while the proteolytic activity of the enzyme is severely inhibited by the presence of copper at all the concentrations tested (as previously reported by us)18–20 and for all the IDE mutants considered, the E1-like activity is barely affected by these metal ions when present in the solution for concentrations up to 20 μM. This striking finding indicates the possibility, in vitro, of diverting the enzyme activity towards the E1-like action by simply using copper concentrations below 20 μM. Remarkably, some of us already demonstrated that under the experimental conditions adopted here copper(ii) ions do not reduce to copper(i).27 Therefore, copper(ii) ions affect IDE activity by direct binding to the protein and not by ROS produced via Fenton chemistry. At this stage, we cannot say that the interaction between copper ions and IDE is significant also in vivo, but, as a dyshomeostasis of copper as well as a decreased IDE proteolytic activity is widely reported in AD, it is easy to speculate that, due to the increased copper levels, a pathological diversion toward the E1-like activity of the enzyme may occur in AD brains. This is particularly relevant in the case of the intracellular activity of IDE. Indeed, it has been reported that Aβ peptides are present in the monomeric form also intracellularly,28 where IDE can contribute to their catabolism. If the dyshomeostasis of copper hinders the intracellular IDE proteolytic activity, then Aβ accumulation might occur, resulting in the deposition of the well-known extracellular amyloid plaques.
Moreover, it is important to highlight that all the IDE mutants investigated, as well as the wt enzyme, are able to produce only diubiquitin molecules, whereas longer polyubiquitin chains are not formed. As it is widely reported that diubiquitin molecules are largely expressed in mammalian cells and represent biologically relevant species, which can be differently recognized by downstream receptor proteins,29 the finding that IDE is unable to produce polyubiquitin chains points out to a very special E1-like role for this enzyme. Indeed, being also aware that IDE is an allosteric modulator of the 20S proteasome and a potential competitor of the 19S,11 it is possible to envisage a very complex role of IDE in the cellular degradation machinery. While copper free IDE is very active for the degradation of amyloidogenic proteins (Aβ, insulin, etc.), the copper bound enzyme shifts its activity toward the production of free diubiquitin molecules, which are only weak inhibitors of the proteasome compared to free polyubiquitin chains.30 Therefore, through direct interaction with the proteasome, as well as the production of diubiquitin molecules, IDE seems to play a very important role in regulating the proteasome activity. Further future in-cell studies could determine the impact that the above reported in vitro biomolecular findings have in vivo.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The financial support from the Italian Ministry of University and Research (MiUR PRIN20157WZM8) and from the Università degli Studi di Catania (Piano della Ricerca di Ateneo 2016-2018) is gratefully acknowledged. Prof. Enrico Rizzarelli is also gratefully acknowledged for his advice and support with the IDE mutants used in this work.
References
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c8mt00288f

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}