
Abstract
Based on the crystal structures of the previously proposed low energy η -P and θ -P, five new phosphorene allotropes were predicted through gene segments recombination method. These five new phosphorene allotropes are confirmed dynamically stable and energetically more favorable than their parents (η -P and θ -P). Especially, the XX-XX type G1-P is confirmed energetically more favorable than most of all the previously proposed phosphorene allotropes, including black phosphorene and blue phosphorene, which is highly expected to be synthesized in future experiment through vapor deposition or epitaxial growth method like blue β -P. The calculated results also show that such a new promising phosphorene allotrope G1-P is a potential candidate for application in nano-electronics according to its middle band gap of about 1.491 eV from DFT-HSE06 calculation.
Similar content being viewed by others

Introduction
Black phosphorene (α -P)1,2, a new atomic thin two-dimensional material, was successfully exfoliated from its three-dimensional counterpart black phosphorus through mechanical method in 2014. As a new-star in two dimensional materials family, black phosphorene is considered as a formidable competitor to graphene and other two-dimensional materials for application in nano-electronic fields due to its significant band gap3,4 and high carrier mobility2. Thus, synthesis of such a new material with low-cost and high-yielding deposition methods, such as chemical or physical vapor deposition, is highly expected.
Usually, the possible crystal structure for a quasi two dimensional phosphorene synthesized from deposition methods always depends on the substrate, pressure, temperature and other experimental conditions. Theoretically, many potential quasi two dimensional phosphorene allotropes have been proposed in the past few years, including the black α -P1,2, blue β -P5,6, γ-P7,δ -P7, θ0-P8 and red phosphorene9 in a full 6–6 ring atomic thin layer, the diatomic thin layers η -P and θ -P with pentagons10, as well as some other atomic thin layers with 4–8, 5–7, 5–8 or 3–12 type topological characteristics7,10,11. In these phosphorene allotropes, the most stable five ones are stirrup black α -P, tricycle red phosphorene, diatomic thin θ -P, chair blue β -P and diatomic thin η -P, in which that the third stable chair blue phosphorene have been synthesized recently12.
Very recently, twenty one quasi two dimensional porous phosphorene allotropes were proposed13,14 through topological modeling method and investigated by first principles calculations. These new potential phosphorene allotropes showing turnable energy band gaps, which provide us many potential structural candidates to understand future experiment. In this letter, we apply the gene segments recombination method15,16 to the previously proposed diatomic thin layers θ -P (provide us XX gene segment) and η -P (provide us XY gene segment) and find five new structural stable phosphorene boys (XX-XY or XY-XY) and girls (XX-XX) with distinct and fascinating two dimensional topology patterns (See in Fig. 1). Density functional theory (DFT) based first-principles method is employed to investigate the structures, energetic stabilities, dynamical stabilities and electronic properties of these five new possible phosphorene allotropes. Our results show that these five new phosphorene allotropes are dynamically stable and three of them are more favorable than their parents in energy. Especially, the XX-XX type girl G1-P is confirmed energetically more favorable than all the previously proposed phosphorene allotropes, including its mother θ -P and the experimentally viable black α -P and blue β -P. Such a result suggests that G1-P is a promising new two dimensional material with high probability to be synthesized in future vapor deposition experiments. The results also show that G1-P is a potential candidate for application in nano-electronics according to its middle band gap of 1.491 eV from DFT-HSE06 calculation.

Top views and corresponding topological patterns of diatomic thin η -P, θ -P and their descendants G1, G2, G3, B1 and B2. The two gene segments XX and XY abstracted form θ -P and η -P, respectively, are also shown here.
Results and Discussion
Crystal structures
As shown in Fig. 1 and Fig. S1, two structural segments can be abstracted from the previously proposed low energy diatomic thin phosphorene allotropes θ -P and η -P. They are named as XY and XX, respectively, according to their structural characteristics. We can see that the XY (XX) gene is consisted of X-Y (X-X) stacked bilayer (we classify θ -P, η -P and their posterities to category of bilayers) of armchair phosphorus chains connecting by inter-chain phosphorus atoms (highlighted as red balls). Based on gene segments XX and XY, we propose five new phosphorene allotropes with remarkable energetic stabilities. In fact, infinite allotropes can be constructed by XX and XY gene segments, but we considered only the situations containing two gene segments per unit cell here. We define those containing only XX gene in their bodies as female girls and name them as G1, G2 and G3. Those containing XY gene are correspondingly defined as male boys and they are named as B1 and B2 in our work. In Fig. 1, the top views of G1, G2, G3, B1, B2 and their parents θ -P and η -P are shown.
From Fig. 1, we can also see that the stacking type between two adjacent XX and/or XY gene segments in each phosphorene allotrope can form individual tiling pattern, which provides us helpful topology characteristics to distinguish them. Only seven staking types are considered in present work, they are θ -P, G1, G2 and G3 of XX-XX, η -P and B2 of XX-XY and B1 of XY-XY. Other stacking types have also been considered in testing task but they possess relatively high energy, and thus are not considered in our present work. These five new allotropes contain two gene segments in their crystalline lattice, which are different to their parents those contain only one gene segment in their crystalline cell. Detail structural information, such as crystalline view from different directions, symmetry group, lattice constants and atomic positions, of these phosphorene allotropes are prepared in the Supplementary file. One can reproduces these phosphorene crystals according to their crystalline information to further study their structures or investigate their other physical properties.
Energetic and dynamic stabilities
To evaluate the relatively energetic stabilities between these phosphorene allotropes, we calculated their total energies relative to that of the black α -P. All previously proposed phosphorene allotropes and some other possible structures (mutations from the previously proposed ones) were also considered in for comparison. The results are classified according to their structural characteristics and plotted in Fig. 2 Our calculations show that the total energy of θ -P and β -P relative to α -P are 15 meV/atom and 19 meV/atom, respectively, which are good consistent with those reported in previous work10 and are reliable according to our testing results for cut off energy and K-mesh (See Fig. S2). From Fig. 2, we can see that some previously proposed phosphorene allotropes (marked as red five-pointed stars) are not the most stable one in their corresponding categories. For example, in single-layered 5–7, 3–12 and 5–8, we find some more favorable candidates. They are marked as black five-pointed stars in Fig. 2 and please see them for more detail in the Supplementary Figures S3–S7 and Tables S2–S7.

The total energy of α -P is set to be zero as reference. From these results, we can see that G1 is energetically more stable than most of all the previously proposed 2D phosphorene allotropes.
As displayed in Fig. 2 in the category of bilayers, the five new allotropes (G1, G2, G3, B1 and B2) constructed through gene segment recombination in our present work show remarkable stability. The total energies of G1, G2, G3, B1, B2, η -P and θ -P relative to α -P are −9 meV/atom, 14 meV/atom, 41 meV/atom, 17 meV/atom, 34 meV/atom, 38 meV/atom and 15 meV/atom, respectively. We can see that G1 is more favorable than its parents and G2, G3, B1 as well as B2 are energetically comparable to their parents. Especially, the XX-XX type G1 is more favorable than most of all (except the multilayered phosphorene bilayer and Hittorfene17) the previously proposed two dimensional phosphorene allotropes, including the experimentally achieved black α -P and blue β -P. Although the energy differences between some phosphorene structures are lie in DFT undistinguishable range (level in meV), we still believe that we have found a new phosphorene allotrope G1 with excellent stability more favorable than black α -P, according to our testing results (see in Supplementary Fig. S2) and the fact that the bilayer black α -P and the 3D black phosphorus are more favorable than single layer black α -P due to the impressive inter-layer vdW interaction. We understand such a remarkable energetic stability of XX-XX type G1 is due to its proper staking manner between the two adjacent XX gene chains, in which the inter-chains vdW interactions cause a sizable energy reduction. Such a suppose can also be proofed in our resent work18 about assembling of the 1D phosphorus nanotubes19 into 2D planar structures (PNT(θ,τ)) in different stack manners, which will cause remarkable energy release and result in different stability according to the stacking manner. As listed in Table 1, our HSE06 results also confirm such an energy sequence in these phosphorene allotropes and the fact that XX-XX type G1 is more favorable than black α -P. We believe that there are still many other new forms of phosphorene allotropes will be predicted more favorable than black α -P and G1 in future, such as the one called as Hittorfene proposed by G. Schusteritsch very recently17 and those phosphorus nanotube arrays PNT(θ, τ) proposed by our group18. Further theoretical and experimental efforts are expected to be paid on searching for them and synthesizing them19,20.
From the view of thermodynamics, low energy generally means high probability to be synthesized in experiments if the system is dynamically possible. The XX-XX type G1 with remarkable stability exceeding black α -P is expected to be synthesized in future vapor deposition method. We then care about the dynamical stabilities of these five new phosphorene allotropes to confirm the possibility of to be synthesized. We evaluate their dynamical stability through simulate their vibrational property. As shown in Fig. 3(a), the phonon band structure of G1 phosphorene is free of soft modes associated with structural instabilities. We have also checked the whole Brillouin Zone and find no any imaginary states in its phonon density of states (See Fig. S9). Such results show that allotrope G1 is dynamically stable. The dynamical stabilities of the other four new phosphorene allotropes (G2, G3, B1 and B2) are also confirmed positive according to their phonon band structures and phonon density of states as shown in Supplementary Figs S8 and S9. To confirm the thermal stability of these five new phosphorene allotropes, we have also performed Born-Oppenheimer molecular dynamics (BOMD) simulation. The corresponding results shown in Figs S11 and S12 indicate that these five new phosphorene allotropes can keep intact at 300 K. In view of the fact that θ -P and η -P can stable even at 700 K10, we believe that their transformers G1, G2, G3, B1 and B2 are still intact at more higher temperature.

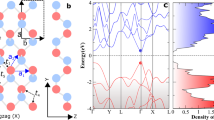
Phonon band structure (a) and electron band structure (b) of G1 calculated form both DFT (black solid line) and HSE06 (blue solid line) methods.
Electronic properties and Raman shift
We then care about the fundamental electronic property of such a promising new phosphorene allotrope G1. The calculated band structure of allotrope G1 is investigated in both DFT and HSE06 methods. As shown in Fig. 3(b), we can see that allotrope G1 is an indirect band gap semiconductor with band gap of 1.491 eV in HSE06 level, which is slightly lower than that of black α -P. Our HSE06 calculated band gap for black α -P is 1.533 eV, which is good consistent with those reported in previous reports13,17,21. Such a promising new phosphorene G1 with middle band gap is a good candidate for application in nano-electronics. The band structures of other four allotropes (η -P, θ -P, G2, G3, B1 and B2) are also investigated by both DFT and HSE06 methods and prepared as Supplementary in Fig. S9. From these results, we can see that all of these new phosphorene allotropes are indirect band gap semiconductors with middle band gaps, which are proper for semiconductor application.
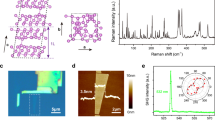
Finally, we simulated the Raman shift spectra of these five new phosphorene allotropes and compared them with those of their parents and black α -P. These Raman shift results are helpful in experiment to differentiate them from each other. As shown in Fig. 4(a), the black solid line and the red solid line are the Raman shift results of α -P from previous experiment1 and present simulation, respectively. We can see that the simulated result matches well with the experimental result1, which confirms that our method is reliable. The simulated Raman spectra of the previously proposed θ -P and η -P are plotted in Fig. 4(b) and (c), respectively, for comparison. We can see that they are very different form that of the black α-P, which indicates that we can easily distinct them out from black α -P through measuring their Raman spectra in experiment if they are synthesized. The Raman shift results of G1, G2, G3, B1 and B2 are plotted in Fig. 4(d–h), respectively. They are very different from those of their parents (θ -P and η -P) and black α -P, and also different from each other. These results provide useful data for experimentally differentiating these potential quasi two dimensional phosphorene allotropes in future.

Raman Shift spectra of the previously proposed black α -P (a), θ -P (b) and η -P, as well as the new G1 (d), G2 (e), G3 (f), B1 (g) and B2 (h). The peak height is in the logarithmic scale and it is smeared by Lorentzian function with a 2 cm−1 width.
Conclusions
In summary, gene segments recombination method was applied to recently proposed diatomic thin layers η -P and θ -P and five new structural stable phosphorene boys (XX-XY or XY-XY) and girls (XX-XX) with distinct and fascinating two dimensional topology patterns were proposed to extend phosphorene allotropes family. Our first-principles calculation results show that these five new phosphorene allotropes are dynamically stable and show remarkable energetic stability. The XX-XX type girl G1-P is confirmed energetically more favorable than most of all the previously proposed phosphorene allotropes, including the experimentally achieved black α -P and blue β -P. Such a result suggests that G1-P is a promising new two dimensional material with high probability to be synthesized in future vapor deposition experiments, which is a potential candidate for application in nano-electronics according to its middle band gap of 1.491 eV from DFT-HSE06 calculation. The experimentally detectable Raman shift properties of these five new phosphorene allotropes are investigated and compared with those of their parents and black α -P, which are very useful for future distinguish them from each other in future experiment.
Methods
Our calculations of structural optimization and properties investigations are carried out by using the density functional theory (DFT) within generalized gradient approximations (GGA)22 as implemented in Vienna ab initio simulation package (VASP)23,24. The interactions between nucleus and the 3s23p3 valence electrons of phosphorus atoms are described by the projector augmented wave (PAW) method25,26. To ensure the accuracy of our calculations, a plane-wave basis with a cutoff energy of 500 eV is used to expand the wave functions and the Brillouin Zone (BZ) sample meshes are set to be dense enough (less than 0.21 Å−1) for each system considered in present work (these settings are set according the convergence test for some important parameters based on the black α -P and G1-P). The structures of these five new phosphorene allotropes and some other reference systems considered in present work are fully optimized up to the residual force on every atom less than 0.001 eV/Å. In such a structural optimization process, the optimized exchange van der Waals functional (optB88-vdW)27,28 is applied to take into account van der Waals interactions. Especially, to gain more reasonable energy band gap of these phosphorene allotropes, the hybrid functional method (HSE06)29 is considered in the processes of properties investigations after structural optimization. We also simulated the vibrational properties of the five new phosphorene allotropes proposed in our present work through the PHONON package30 with the forces calculated from VASP to confirm their dynamical stabilities. For the purpose of providing experimentally detectable property to differentiate these possible phosphorene allotropes, we simulated their Raman shift through CASTEP software31.
Additional Information
How to cite this article: He, C. et al. Five low energy phosphorene allotropes constructed through gene segments recombination. Sci. Rep. 7, 46431; doi: 10.1038/srep46431 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Liu, H. et al. Phosphorene: An Unexplored 2D Semiconductor with a High Hole Mobility ACS Nano 8, 4033 (2014).
Li, L. et al. Black phosphorus field-effect transistors Nat . Nano 5, 372 (2014).
Liu, Y., Xu, F., Zhang, Z., Penev, E. S. & Yakobson, B. I. Two-dimensional mono-elemental semiconductor with electronically inactive defects: the case of phosphorus Nano lett. 14, 6782 (2014).
Liang, L. et al. Electronic bandgap and edge reconstruction in phosphorene materials Nano Lett. 14, 6400 (2014).
Zhu, Z. & Tománek, D. Semiconducting Layered Blue Phosphorus: A Computational Study Phys. Rev. Lett. 112, 176802 (2014).
Boulfelfel, S. E., Seifert, G., Grin, Y. & Leoni, S. Squeezing lone pairs: The A17 to A7 pressure-induced phase transition in black phosphorus Phys. Rev. B. 85, 014110 (2012).
Guan, J., Zhu, Z. & Tománek, Phase Coexistence and Metal-Insulator Transition in Few-Layer Phosphorene: A Computational Study Phys. Rev. Lett. 113, 046804 (2014).
Guan, J., Zhu, Z. & Tománek, D. Tiling Phosphorene ACS Nano. 8, 12763 (2014).
Zhao, T. et al. J. X. A new phase of phosphorus: the missed tricycle type red phosphorene J. Phys.: Condens. Matter. 27, 265301 (2015).
Wu, M., Fu, H., Zhou, L., Yao, K. & Zeng, X. C. Nine New Phosphorene Polymorphs with Non-Honeycomb Structures: A Much Extended Family Nano lett. 15, 3557 (2015).
Yu, G., Jiang, L. & Zheng, Y. Two-dimensional Kagome phosphorus and its edge magnetism: a density functional theory study J. Phys.: Condens. Matter. 27, 255006 (2015).
Zhang, J. L. et al. Epitaxial Growth of Single Layer Blue Phosphorus: A New Phase of Two-Dimensional Phosphorus Nano Lett. 16, 4903 (2016).
Zhuo, Z., Wu, X. & Yang, J. Two-Dimensional Phosphorous Porous Polymorphs with Tunable Band Gaps J. Am. Chem. Soc. 138, 7091 (2016).
Xu, M., He, C., Zhang, C., Tang, C. & Zhong, J. X. First-principles prediction of a novel hexagonal phosphorene allotrope Phys. Solidi. Status.-RRL 10, 563 (2016).
Zhou, R. L. & Zeng, X. C. Polymorphic phases of sp3-hybridized carbon under cold compression J. Am. Chem. Soc. 134, 7530 (2012).
He, C. et al. New superhard carbon phases between graphite and diamond Solid. State. Commun. 181, 24 (2014).
Schusteritsch, G., Uhrin, M. & Pickard, C. J. Single-Layered Hittorf’s Phosphorus: A Wide-Bandgap High Mobility 2D Material Nano. Lett. 16, 2975 (2016).
Li, Z. Q. et al. New allotropes of phosphorene with remarkable stability and intrinsic piezoelectricity arXiv:1701.05075.
Liu, D., Guan, J., Jiang, J. & Tománek, D. Unusually stable helical coil allotrope of phosphorus Nano. Lett. 16, 7865 (2016).
Zhang, J. Y. et al. Assembly of Ring-Shaped Phosphorus within Carbon Nanotube Nanoreactors Angew. Chem. 1, 129 (2017).
Qiao, J., Kong, X., Hu, Z. X., Yang, F. & Ji, W. High-mobility transport anisotropy and linear dichroism in few-layer black phosphorus Nat. Commun. 5, 4475 (2014).
Perdew, J. P. & Yue, W. Accurate and simple density functional for the electronic exchange energy: Generalized gradient approximation Phys. Rev. B 33, 8800 (1986).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set Phys. Rev. B 54, 11169 (1996).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set Comput. Mater. Sci. 6, 15 (1996).
Blöchl, P. E. Projector augmented-wave method Phys. Rev. B 50, 17953 (1994).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method Phys. Rev. B 59, 1758 (1999).
Klimes, J., Bowler, D. R. & Michaelides, A. Chemical accuracy for the van der Waals density functional J. Phys.: Cond. Matt. 22, 022201 (2009).
Klimes, J., Bowler, D.R. & Michaelides, A. Van der Waals density functionals applied to solids Phys. Rev. B 83, 195131 (2011).
Heyd, J., Scuseria, G. E. & Ernzerhof, M. Hybrid functionals based on a screened Coulomb potential J. Chem. Phys. 118, 8207 (2003).
Parlinski, K., Li, Z.-Q. & Kawazoe, Y. First-principles determination of the soft mode in cubic ZrO2 Phys. Rev. Lett. 78, 4063 (1997).
Clark, S. J. et al. First principles methods using CASTEP Z. Kristallogr.-Cryst. Mater. 220, 567 (2005).
Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant Nos 11647063, A040204 and 11204261), the National Basic Research Program of China (2012CB921303 and 2015CB921103), the Young Scientists Fund of the National Natural Science Foundation of China (Grant No. 11204260), the Natural Science Foundation of Hunan Province, China (Grant No. 2016JJ3118, the Scientific Research Found of HuNan Provincial Education department (No. 14C1095), and the Program for Changjiang Scholars and Innovative Research Team in University (IRT13093).
Author information
Authors and Affiliations
Contributions
C.Y. He and C. Tang conceived the initial idea of this research. C.Y. He and C.X. Zhang conceived the calculations and collected all data. C.Y. He and J. Li prepared all figures. All the authors participated in the discussions and analyzed the data. C.Y. He and T. Ouyang wrote the manuscript. C. Tang and J.X. Zhong designed and coordinated the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
He, C., Zhang, C., Tang, C. et al. Five low energy phosphorene allotropes constructed through gene segments recombination. Sci Rep 7, 46431 (2017). https://doi.org/10.1038/srep46431
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep46431
This article is cited by
-
Alcohol molecular interaction studies on stair phosphorene nanosheets: a first-principles approach
Structural Chemistry (2021)
-
Interaction Behavior of Cyanogen Fluoride and Chloride Gas Molecules on Red Phosphorene Nanosheet: A DFT Study
Journal of Inorganic and Organometallic Polymers and Materials (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
