
Abstract
Endothelin-1 (ET-1) promotes renal damage during cardiovascular disease; yet, the molecular mechanisms involved remain unknown. Endoplasmic reticulum (ER) stress, triggered by unfolded protein accumulation in the ER, contributes to apoptosis and organ injury. These studies aimed to determine whether the ET-1 system promotes renal ER stress development in response to tunicamycin. ETB deficient (ETB def) or transgenic control (TG-con) rats were used in the presence or absence of ETA receptor antagonism. Tunicamycin treatment similarly increased cortical ER stress markers in both rat genotypes; however, only ETB def rats showed a 14–24 fold increase from baseline for medullary GRP78, sXBP-1, and CHOP. Pre-treatment of TG-con rats with the ETA blocker ABT-627 for 1 week prior to tunicamycin injection significantly reduced the ER stress response in cortex and medulla, and also inhibited renal apoptosis. Pre-treatment with ABT-627 failed to decrease renal ER stress and apoptosis in ETB def rats. In conclusion, the ET-1 system is important for the development of tunicamycin-induced renal ER stress and apoptosis. ETA receptor activation induces renal ER stress genes and apoptosis, while functional activation of the ETB receptor has protective effects. These results highlight targeting the ETA receptor as a therapeutic approach against ER stress-induced kidney injury.
Similar content being viewed by others


Introduction
Upregulation of the endothelin (ET) system has been reported in a wide range of cardiovascular and renal diseases1,2; however, the exact cellular and molecular mechanisms through which endothelin-1 (ET-1) leads to renal injury are not fully discerned. ET-1 is an endogenous 21 amino acid peptide with strong vasoactive attributes. The effects of ET-1 are mediated by two G protein-coupled receptors: ETA and ETB receptors. Both receptors bind to ET-1 with the same affinity; however, activation of each receptor subtype leads to opposite physiological and pathophysiological effects. For instance, overactivation of ETA receptors in the kidney promotes renal hypertrophy, fibrosis and inflammation. On the other hand, activation of ETB receptors helps to clear ET-1 from the circulation, as well as stimulating Na+and water excretion by inhibition of tubular reabsorption through production of nitric oxide and prostaglandins3. Within the renal vasculature, the majority of ETB receptors are located on the endothelium and smooth muscle of the efferent arteriole, whereas ETA receptors are predominant in the afferent arteriole vascular smooth muscle. The distribution of these receptors along the nephron is also distinct: ETB receptors are abundant in cortical and inner medullary tubules, with both ETA and ETB receptors present in tubules of the outer medulla3.
Recently, endoplasmic reticulum (ER) stress has been highlighted as a mechanism involved in renal apoptosis and renal injury4. ER stress is a type of cellular stress that results from the accumulation of unfolded proteins in the ER. In order to maintain homeostasis, the cell activates the adaptive unfolded protein response (UPR). The ER chaperone protein glucose-regulated protein 78 (GRP78; considered to be the master regulator of the ER stress response)5, recognizes the unfolded proteins, physically binds to these proteins, and initializes the 3 parallel arms of the UPR. This leads to the activation of spliced X box-binding protein-1 (sXBP-1), activating transcription factor-4 (ATF-4) and ATF-6, which translocate to the nucleus to temporarily stop further protein transcription and translation; hence, the adaptive UPR gains time for the ER to fold the accumulating misfolded proteins. In case of severe or prolonged ER stress, the cell activates the apoptotic UPR by upregulating expression of the transcription factor CCAAT-enhancer-binding protein homologous protein (CHOP) and caspase-12, leading to cell death via apoptosis and eventually inducing organ damage5.
Evidence in the literature demonstrates an important role of ER stress in the development of acute kidney injury (AKI) in humans and in animal models of this disease6,7,8. Furthermore, both ET-1 and ER stress are upregulated in renal diseases such as contrast-induced acute kidney injury9,10, ischemia/reperfusion injury11,12, septic shock-induced acute kidney injury13,14, and diabetic nephropathy15,16, suggesting that overactivation of the ET-1 system may lead to induction of the renal ER stress response. Consistent with this possibility, induction of the UPR by ET-1 has been shown in pulmonary aortic smooth muscle cells17 and placental tissue18. On the other hand, other authors suggest that activation of the ER stress response mediates ET-1 release from aortic endothelial cells during endothelial dysfunction19.
It has been reported that renal injury is preceded by tubular apoptosis and loss of nephrons20, and several vasoactive peptides have been implicated in the regulation of cellular apoptosis. However, there are contradictory reports in the literature regarding the role that ET-1 plays in the development of apoptosis and renal injury, with some reports indicating that ET-1 induces cellular apoptosis21,22 and others suggesting the opposite23,24,25.
The present studies aimed to clarify the role of the ET-1 system in the development of renal ER stress and apoptosis utilizing the ER stress inducer tunicamycin. Similar to other agents mediating kidney damage, such as cisplatin or adriamycin, tunicamycin is commonly used to model antibiotic-mediated acute kidney injury26,27,28. Tunicamycin induces ER stress by inhibiting protein glycosylation and preventing correct protein folding, which results in protein accumulation in the ER and activation of the ER stress response29. We hypothesized that the ET-1 system contributes to the development of tunicamycin-induced renal ER stress and apoptosis. Through genetic and pharmacological approaches, we demonstrate that activation of the ETA receptor is important for the induction of apoptosis and the ER stress response in the kidney early in the progression of tunicamycin-induced injury. We also demonstrate the protective role of functioning ETB receptors against tunicamycin-induced renal ER stress and apoptosis.
Results
Assessment of the systemic and renal ET-1 system in response to tunicamycin
To study the role of ET-1 receptors in the development of renal ER stress and apoptosis, transgenic control and ETB deficient rats (TG-con and ETB def rats) were treated with a single i.p. injection of tunicamycin (2 μg/g body weight) or saline and studied 24 hours later. The ETB def rats have a natural occurring mutation of the ETB receptor that renders this receptor dysfunctional30. As shown in Fig. 1a and b, ET-1 excretion and plasma ET-1 levels were not significantly changed by tunicamycin in either genotype. Moreover, treatment with tunicamycin did not significantly change mRNA expression of pre-pro-ET-1 in renal cortex or outer medulla of either genotype (Fig. 1c). Thus, tunicamycin does not alter circulating or renal ET-1 levels in ETB def or TG-con rats.

(a) Urinary excretion of ET-1 in TG-con and ETB def rats treated with saline or tunicamycin; n = 4–5/group. (b) Plasma ET-1 levels in TG-con and ETB def rats treated with saline or tunicamycin; *P < 0.05 vs. TG-con + same treatment; n = 4–5/group. (c) Relative mRNA expression of pre-pro-ET-1 in renal cortex and outer medulla from TG-con and ETB def rats after treatment with saline or tunicamycin; n = 4–5/group. RNA expression was normalized to same genotype + saline. Statistical significance was determined by two-way ANOVA with Tukey post hoc test.
Assessment of tunicamycin-induced ER stress markers in the kidney
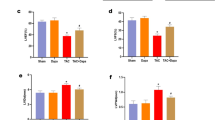
To explore the potential involvement of ETB receptors in the development of ER stress, mRNA expression of ER stress markers was measured by qRT-PCR in renal cortex and outer medulla of TG-con and ETB def rats treated with saline or tunicamycin (Fig. 2). Preliminary studies showed no changes in expression of ER stress markers in renal inner medulla; therefore, the present studies focused solely on the cortex and outer medulla. TG-con and ETB def rats treated with saline did not differ with regard to mRNA expression of ER stress markers in cortex or outer medulla (Fig. 2). In the renal cortex, TG-con rats responded to the tunicamycin challenge with an 11-fold increase in expression of GRP78 and a 7-fold increase in sXBP-1 expression (n = 6–9/group; Fig. 2a). In addition to upregulation of these two markers, tunicamycin treatment of ETB def rats significantly increased expression of three additional markers in this region of the kidney: ATF-6, CHOP, and caspase-12 (with fold increases between 3 and 31; n = 6–9/group; P < 0.05). Similar to mRNA expression, prominent GRP78 immunostaining was evident in distal nephron segments within the renal cortex of tunicamycin-treated rats of both genotypes (Fig. 3a and c). CHOP immunostaining in the renal cortex of ETB def rats appear most prominent in distal nephron segments and not as prominent in TG-con, although this difference was not significant (Fig. 3b and d).

Relative mRNA expression of ER stress markers in renal cortex (a) and outer medulla (b) from TG-con and ETB def rats after treatment with saline or tunicamycin. †P < 0.05 vs. saline (same genotype); n = 6–9/group. RNA expression was normalized to same genotype + saline. Statistical significance was determined by two-way ANOVA with Tukey post hoc test.

Representative images of protein expression of GRP78 (a) and CHOP (b) in renal cortex of TG-con and ETB def rats treated with saline or tunicamycin. Bar = 50 μm. (c) Percentage of cortex stained positive for GRP78 (n = 4–5/group). (d) Percentage of cortex stained positive for CHOP (n = 4–5/group).
The outer medulla exhibited tunicamycin-induced changes in mRNA expression of ER stress proteins only in ETB def rats. These animals responded to the tunicamycin challenge with significant increases in outer medullary mRNA expression of GRP78 (14-fold), sXBP-1 (10-fold) and CHOP (24-fold) (n = 6–9/group; P < 0.05; Fig. 2b), with no change in expression evident in TG-con rats. The protective effects of the ETB receptor in the outer medulla were also evident at the protein level. As shown in Fig. 4a and c, GRP78 immunostaining was significantly elevated in a subset of outer medullary tubular segments in tunicamycin-treated ETB def rats. Outer medullary CHOP immunostaining tended to be increased with tunicamycin treatment in this genotype (Fig. 4b), however, it was not statistically different from the saline-treated group (Fig. 4d). Tunicamycin did not markedly influence outer medullary GRP78 or CHOP immunostaining in TG-con rats. These results highlight the possible protective effect of the ETB receptor against ER stress in tubular segments located in the outer medulla, as absence of this receptor led to development of tunicamycin-induced ER stress in this area of the kidney.

Representative images of protein expression of GRP78 (a) and CHOP (b) in renal outer medulla of TG-con and ETB def rats treated with saline or tunicamycin. Bar = 50 μm. (c) Percentage of cortex stained positive for GRP78 (n = 4–5/group). (d) Percentage of cortex stained positive for CHOP (n = 4–5/group). †P < 0.05 vs. same genotype + saline; *P < 0.05 vs. TG-con + tunicamycin. Statistical significance was determined by two-way ANOVA with Tukey post hoc test.
To assess the role of the ETA receptor in the development of renal ER stress, TG-con and ETB def rats were pre-treated with the specific ETA antagonist ABT-627 (5 mg/kg/day via the drinking water) or left untreated (vehicle), for one week prior to tunicamycin administration. Pre-treatment with ABT-627 significantly blunted cortical and outer medullary expression of GRP78 and CHOP in TG-con rats (GRP78 decreased by 69% in cortex and 78% in outer medulla; CHOP decreased by 77% in cortex and 86% in outer medulla; n = 6–10/group; P < 0.05). In addition, pre-treatment with ABT-627 significantly blunted expression of sXBP-1 and caspase-12 in cortex (decreased by 77% and 82%, respectively; n = 6–10/group; P < 0.05), and ATF-4 in outer medulla of TG-con (decreased by 75%; n = 6–10/group; P < 0.05). Similar trends were apparent regarding ATF-6, although not reaching statistical significance (Fig. 5). These results indicate that activation of the ETA receptor is important for the development of tunicamycin-induced ER stress in the kidney.

Effects of ETA receptor antagonist (ABT-627) on mRNA expression of ER stress markers in renal cortex (a) and outer medulla (b) in tunicamycin-treated TG-con and ETB def rats. †P < 0.05 vs. same genotype + saline; ‡P < 0.05 vs. same genotype + ABT-627 + tunicamycin; *P < 0.05 vs. TG-con + ABT-627 + tunicamycin. n = 6–10/group. RNA expression was normalized to same genotype + saline. Statistical significance was determined by two-way ANOVA with Tukey post hoc test.
In contrast to TG-con rats, pre-treatment with ABT-627 did not protect ETB def rats from tunicamycin-induced renal ER stress, as mRNA expression of ER stress markers remained elevated in both the cortex and outer medulla. Expression of caspase-12 was also significantly elevated in these animals in response to the tunicamycin challenge. These results further support the protective role of the ETB receptor against the development of renal ER stress in response to tunicamycin, as the absence of functional ETB receptors leads to increased expression of ER stress markers in both areas of the kidney regardless of ETA receptor status.
Assessment of tunicamycin-induced renal apoptosis
To assess the role of the ETA receptors in the development of tunicamycin-induced renal apoptosis, TUNEL assay was performed in kidneys from TG-con and ETB def rats receiving ABT-627 via the drinking water for one week prior to the injection of tunicamycin. As indicated in Fig. 6, TUNEL-positive cells were evident both in the cortex and, to a greater extent, in the outer medulla 24 hours after tunicamycin administration to TG-con and ETB def rats. Pre-treatment of TG-con rats with the ETA receptor antagonist almost completely obliterated the tunicamycin-induced apoptosis evident in the renal cortex (decreasing from 13.5 ± 1.6 to 1.3 ± 0.4 TUNEL-positive cells/field; n = 5–6/group; P < 0.05; Fig. 6c) and outer medulla (decreasing from 30.2 ± 2.7 to 1.6 ± 0.4 TUNEL-positive cells/field; n = 5–6/group; P < 0.05; Fig. 6c). In contrast, ABT-627 failed to prevent the development of tunicamycin-induced renal apoptosis in ETB def rats, in both cortex and medulla (17.6 ± 2.0 TUNEL-positive cells/field in cortex and 39.0 ± 4.4 TUNEL-positive cells/field in medulla), further highlighting the important role of the ETB receptor in protecting against the development of renal apoptosis. Closer examination of these images at high magnification (Fig. 7) reveals that the TUNEL-positive cells within the renal tissue are not tubular cells, but interstitial cells located between tubules and/or near renal vasa recta.

Effects of tunicamycin on renal apoptosis in renal cortex (a) and outer medulla (b) of TG-con and ETB def rats pre-treated with vehicle or ABT-627 (apoptosis detected by TUNEL assay). Bar = 50 μm. (c) Quantification of TUNEL positive cells in renal cortex and outer medulla. †P < 0.05 vs. same genotype + saline; ‡P < 0.05 vs. same genotype + ABT-627 + tunicamycin; *P < 0.05 vs. TG-con + ABT-627 + tunicamycin. n = 5–6/group. Statistical significance was determined by two-way ANOVA with Tukey post hoc test.

Representative high magnification images of interstitial apoptotic cells in cortex and outer medulla of an ETB def rat treated with ABT-627 + tunicamycin. (a) Glomerular area, (b) cortical tubular area, and (c) outer medullary vasa recta area. Blue dashed lines outline tubules and red dashed lines outline vessels. Bar = 20 μm.
Assessment of renal injury and renal function in response to tunicamycin
To assess whether the acute treatment with tunicamycin increases renal injury, we determined urinary albumin excretion, histological assessments of injury, and renal inflammatory cell numbers. Albumin excretion, a sensitive marker of renal injury, was significantly elevated in both the TG-con and ETB def animals (Fig. 8a). Pre-treatment with ABT-627 prevented tunicamycin-induced increases in albumin excretion in TG-con rats. These effects on albumin excretion were absent in ETB def rats, suggesting that the presence of a functional ETB receptor is important to prevent the development of albuminuria in response to treatment with tunicamycin (Fig. 8a). Immunostaining for ED-1 and CD3 was utilized to assess infiltration of macrophages and T-lymphocytes, respectively, in ETB def and TG-con rats. Numbers of macrophages and T-lymphocytes did not differ between kidneys from ETB def or TG-con rats treated with saline or tunicamycin in any of the studied renal regions (Supplementary Figure 1; n = 5/group). Examination of renal histology demonstrated no differences in glomerular sclerosis, interstitial fibrosis or proximal tubule brush border thickness after treatment of both genotypes with tunicamycin (data not shown). However, we observed that tunicamycin led to vasa recta injury in the outer medulla of TG-con and ETB def rats, as indicated by stronger periodic acid Schiff (PAS) staining when compared to the same genotypes treated with saline (Supplementary Figure 2).

(a) Albumin excretion in TG-con and ETB def rats treated with saline, tunicamycin or pre-treated with ABT-627 for one week and then given tunicamycin; n = 4–5/group. (b) Plasma creatinine levels in TG-con and ETB def rats treated with saline, tunicamycin or pre-treated with ABT-627 for one week and then given tunicamycin; n = 6–7/group. (c) Plasma BUN levels in TG-con and ETB def rats treated with saline, tunicamycin or pre-treated with ABT-627 for one week and then given tunicamycin; n = 3–5/group. †P < 0.05 vs. TG-con + tunicamycin. †P < 0.05 vs. same genotype + saline; ‡P < 0.05 vs. same genotype + ABT-627 + tunicamycin; *P < 0.05 vs. TG-con + ABT-627 + tunicamycin. Statistical significance was determined by two-way ANOVA with Tukey post hoc test.
In addition, we assessed whether the acute treatment with tunicamycin alters renal function by measuring plasma creatinine, creatinine clearance, and plasma blood urea nitrogen (BUN). Creatinine clearance was unchanged (TG-con vs. ETB def; saline: 2.0 ± 0.3 vs. 2.3 ± 0.2 ml/min, tunicamycin: 2.7 ± 0.2 vs. 2.6 ± 0.5 ml/min), as well as plasma creatinine levels or plasma blood urea nitrogen (BUN) levels in the experimental animals (Fig. 8b and c). Of note, pre-treatment with ABT-627 did not lead to changes in any of these measures of renal function (Fig. 8b and c).
Discussion
The present study demonstrates that the ETA and ETB receptors play opposite roles in the development of ER stress and apoptosis in the kidney in response to tunicamycin. On one hand, activation of the ETA receptor is important for tunicamycin-induced ER stress and apoptosis in the kidney as well as increased albumin excretion, and, on the other hand, activation of the ETB receptor ameliorates and is necessary for the protection against the renal injury by inhibiting ER stress and renal apoptosis. Despite extensive evidence supporting the role of ET-1 and its receptors in the pathophysiology of kidney disease, the cellular and molecular mechanisms by which this vasoactive peptide mediates the development of renal injury remain unknown. In this paper we demonstrate that the ET-1 system is involved in the development of renal ER stress and apoptosis as well as albuminuria induced by tunicamycin.
The results of the present study indicate that ETA receptor activation is important for the development of tunicamycin-induced ER stress in the kidney. Specifically, pharmacological blockade of ETA receptors with ABT-627 dramatically decreased the expression of ER stress markers in both renal cortex and outer medulla of tunicamycin-treated TG-con rats. Our results agree with previous reports that the ET-1 system is capable of inducing ER stress in cultured pulmonary aortic smooth muscle cells17 or placental tissue during pre-eclampsia18. Activation of the ETA receptor has been shown to stimulate renal fibrosis, inflammation and increase albumin permeability31,32,33, hallmarks of renal injury which has been linked to ER stress. For instance, it has been reported that inhibition of the UPR response in a well-known model of kidney fibrosis, the unilateral urethral obstruction model, leads to amelioration of fibrosis34, and similarly, the three arms of the UPR have been shown to activate the central inflammatory transcription factor, NFκB35. At this point we are unsure of how activation of the ETA receptor leads to upregulation of ER stress pathways; however, it has been widely reported that activation of this receptor leads to the production of superoxide by stimulation of the NADPH oxidase36. It is also known that oxidative stress can stimulate the UPR as an adaptive mechanism to preserve cell physiology during renal dysfunction37. Thus, stimulation of oxidative stress could be a possible mechanism by which activation of the ETA receptor may be leading to the development of renal ER stress. Alternatively, glycosylation of endothelin receptors is important for their function38, thus the inhibition of ETA receptor glycosylation by tunicamycin may be affecting the binding of endothelin and/or the specific post-receptor signaling pathways.
Using the ETB deficient (ETB def) rat as an experimental model, the present study revealed the protective role of this receptor against the development of renal ER stress. ETB deficient rats have dysfunctional ETB receptors due to a natural occurring mutation of this gene. Because complete lack of the ETB receptor results in premature death, these rats were rescued years ago by the re-introduction of the ETB receptor in the neuronal tissue; as a consequence, they express functional ETB receptors only in the nerves, while the rest of the tissues (including the kidneys) have non-functional ETB receptors30. Because of the importance of the ETB receptor in clearing plasma ET-1, ETB def rats present elevated levels of plasma ET-1 and overactivation of ETA receptors30. This phenomenon, in and of itself, is insufficient to provoke ER stress, as expression of ER stress markers did not differ between genotypes in the absence of tunicamycin. However, when presented with a “second hit” of a relatively low dose and a single injection of this ER stress inducer, ETB def rats developed an exaggerated renal ER stress response. This response was especially dramatic in the outer medullary region, where ETB receptors are known to be more abundantly distributed than ETA receptors39,40. The effects of ABT-627 were absent in the ETB def rats, once again highlighting the protective role of the ETB receptor against the development of renal ER stress. These findings indicate that the ETB receptor opposes the pro-ER stress actions of the ETA receptor and, when the ETB receptor is dysfunctional, the unopposed activation of the ETA receptor leads to an exaggerated ER stress response in the kidney. It is well known that activation of the ETB receptor leads to nitric oxide release41,42; thus, upregulation of nitric oxide production may be a possible mechanism through which the ETB receptor protects against ER stress development in the kidney.
Tubular apoptosis and loss of nephrons are known to precede kidney injury20. Different vasoactive peptides have been implicated in the regulation of apoptosis; however, reports in the literature are contradictory regarding the role of ET-1 in this cellular process. Some studies describe pro-apoptotic effects of ET-1 in vascular smooth muscle cells21 or in different parts of the kidney like glomeruli, tubules or interstitial cells22. On the other hand, other publications report that ET-1 attenuates apoptosis in fibroblasts25, vascular smooth muscle cells23 and endothelial cells24. The role of ET-1 receptors in apoptosis is also controversial in the literature. Some reports describe pro-apoptotic effects of the ETA receptor in chronic renovascular disease43 and polycystic kidney disease44,45, while others indicate that activation of this receptor promotes cell proliferation and survival during kidney development46, in cardiomyocytes47 or in vascular smooth muscle cells48. Additionally, the loss or inhibition of ETB receptors has been reported as protective against apoptosis in neurons that underwent hypoxia-ischemia49, whereas ETB selective agonists led to decreased apoptosis in rat endothelial cells25 and in tubules from a mouse model of polycystic kidney disease45. Other studies described that the use of an ETB blocker increased apoptosis in rat and human endothelial cells24,50 and in human melanoma lines51.
In addition to effects on the ER stress response, the present study revealed that specific pharmacological blockade of ETA receptor ameliorates tunicamycin-induced renal apoptosis in TG-con rats, while failing to do the same in ETB deficient rats. These results highlight the important role that activation of the ETA receptor has in promoting tunicamycin-induced renal apoptosis. The fact that the renal tubular apoptosis is not diminished by ETA blockade in the ETB deficient rats also emphasizes the protective role of the ETB receptor in opposing the pro-apoptotic effects of the ETA receptor. Interestingly, we found that tubular cells display upregulation of CHOP at the mRNA and protein levels; however, the cells undergoing apoptosis are interstitial cells, rather than the tubular cells. Since tunicamycin treatment did not increase renal infiltration of macrophages or T cells in our acute model, we speculate that the apoptotic cells may be resident immune cells in the peritubular interstitium tissue. Immune cells such as macrophages52 or dendritic cells53 possess ETA and ETB receptors, are responsive to ET-1, and are also able to synthesize and release ET-152,53. Thus, these immune cells may also respond to tunicamycin and activate apoptotic pathways influenced by the ET-1 system. Accelerated macrophage apoptosis induces autoantibody formation and organ damage in lupus nephritis54, mainly through increased apoptotic load in the tissue and decreased apoptotic body clearance. Hence, we hypothesize that resident immune cell apoptosis may be the mechanism that leads to the activation of UPR pathways in the renal tubules in our animal model. Further studies are needed to clarify this point.
Although results of the present study indicate that tunicamycin-induced apoptosis is mediated by the ET-1 system, tunicamycin has also been reported to lead to apoptosis through stimulation of oxidative stress28, among other pathways. Because ETB receptors counteract the oxidative stress induced by activation of ETA receptors36, the renal apoptosis evident in the ETB deficient rats pre-treated with ABT-627 could be due to activation of these alternative pathways by tunicamycin and worsened due to the absence of a functional ETB receptor in these animals.
Finally, these studies also find that this acute tunicamycin treatment induces albuminuria, a sensitive marker of renal injury, in both genotypes but is only ameliorated in the transgenic controls rats with ETA receptor antagonism not in the ETB deficient rats. We documented injury of the vasa recta in both genotypes with the acute tunicamycin treatment. Although other histological measures, such as glomerulosclerosis and tubular fibrosis, were not observed. Further, measures of renal function such as plasma creatinine and BUN, were also not affected by the tunicamycin treatment. These negative findings are most likely due to the acute nature of the experimental protocol.
In conclusion, these findings highlight the potential therapeutic value of specifically targeting the ETA receptor system to prevent the development of antibiotic induced acute renal injury mediated via ER stress and apoptosis. Based on the results presented, we propose that an insult, for instance tunicamycin, stimulates ETA receptors in the tubular epithelium as well as interstitial immune cells, leading to ER stress, apoptosis and, eventually, kidney damage. In this scheme, ETB receptors function as a brake in the system, attenuating the ETA dependent effects on ER stress and apoptosis in the kidney.
Methods
Animal studies
All protocols were conducted in accordance with the Guide for the Care and Use of Laboratory Animals, and were approved by the University of Alabama at Birmingham and Augusta University Institutional Animal Care and Use Committees. These studies utilized 10–12 week old male ETB deficient rats (specifically, DBH-ETB;ETBsl/sl rats) and their DBH-ETB;ETB+/+ transgenic littermates. DBH-ETB;ETBsl/sl rats (ETB def) express the ETB receptor only in adrenergic tissues, under the transcriptional control of the dopamine β-hydroxylase promoter30. In one set of experiments, ETB def rats and their DBH-ETB;ETB+/+ transgenic littermates (TG-con rats) were placed in metabolic cages for 2 days to acclimate and then received a single i.p. injection of tunicamycin (2 μg/g body weight; Sigma-Aldrich, St. Louis, MO) or saline on the third day. Rats were sacrificed 24 h post-injection, and 24 h urine, plasma and kidneys were collected. In a second set of experiments, 10–12 week old male ETB def and TG-con rats were randomized to receive the ETA receptor antagonist atrasentan (ABT-627; 5 mg/kg/day via drinking water; AbbVie Laboratories, North Chicago, IL) or regular water (vehicle) for 1 week prior to a single injection of tunicamycin (2 μg/g body weight, i.p.). Two days before the injection, the rats were placed in metabolic cages, to allow for urine collection before and after tunicamycin treatment. Twenty four hours post-injection, the rats were sacrificed and plasma and kidneys were harvested; renal cortex and outer medulla were isolated and rapidly snap frozen in liquid nitrogen and kept at −80 °C until further analysis.
Quantitative RT-PCR
RNA was extracted from renal tissue using RNeasy mini kit (Qiagen, Valencia, CA) and quantified by spectrophotometric analysis (NanoDrop ND-1000, Thermo Scientific, Waltham, MA). RNA was reverse transcribed using Quantitect Reverse Transcription kit (Qiagen) following manufacturer’s instructions. ER stress primers were synthesized by Integrated DNA Technologies (IDT, Coralville, IA; primer sequences are indicated in Supplemental Table 1 55,56,57) and primers for pre-pro-ET-1 were purchased from Qiagen. GAPDH was used as housekeeping gene. RNA expression was detected with Quantitect SYBR green kit (Qiagen) and using a CFX96 Touch RT-PCR detection system (Bio-Rad, Hercules, CA).
Immunohistochemical analysis
Kidneys were fixed in 4% buffered formalin solution overnight at room temperature, transferred to 70% ethanol for 24 h and paraffin-embedded. Tissues were cut longitudinally into 4 μm-thick sections and mounted on Superfrost slides. Tissue sections were stained with primary antibodies specific for CHOP (1:50; Novus Biologicals, Littleton, CO), GRP78 (1:3,000; Abcam, Cambridge, MA), CD3 (1:400; Abcam), and ED-1 (1:100; Bio-Rad), and detected with polymer conjugated secondary antibody (Biocare Medical, Concord, CA).
Whole kidney scans (100x magnification) were obtained using a scanning microscope fitted with a DP73 camera (Olympus America, Melville, NY), and Metamorph imaging software (Molecular Devices, Sunnyvale, CA) was used to quantify GRP78 and CHOP immunostaining. The cortical and outer medullary areas of each kidney image were outlined using Metamorph software and the amount of positive stain for each antibody was obtained. Data are expressed as the percentage of area of the kidney (cortex or outer medulla) positively stained for GRP78 or CHOP (n = 5/group).
Quantification of renal T-lymphocyte and macrophage infiltration was performed by blindly counting 10 microscopic fields (400 × 400 μm, 200× magnification) in each kidney region (cortex and outer medulla). The numbers are reported as average of the counts in the 10 fields per kidney region.
Histological analysis
Renal structures were visualized with periodic acid Schiff (PAS), trichrome blue, hematoxylin and eosin, and picrosirius red stains using bright-field microscopy (Olympus BX40; Olympus America). Images were obtained with a digital camera (Olympus DP12; Olympus America). Renal damage was evaluated by assessing glomerulosclerosis, interstitial fibrosis, proximal tubule brush border thickness and vasa recta integrity in a blinded manner. For assessing glomerulosclerosis, ten glomeruli per kidney slide were evaluated and each received a glomerulosclerosis score of 1 = 25%, 2 = 50%, 3 = 75%, or 4 = 100%. Scoring of the degree of thickening of vasa recta was performed by using a presence/absence scale, where a score of 0 indicates no thickening present and 1 means presence of thickened vasa recta. Ten vasa recta bundles per experimental animal were scored, with 5 animals per experimental group analyzed. Data are presented as average of those scores per experimental group.
Plasma and urinary ET-1
Levels of ET-1 in undiluted samples of plasma and urine were determined by a chemiluminescent assay (Human ET-1 QuantiGlo kit, R&D Systems, Minneapolis, MN).
Renal function and renal injury marker determination
Plasma and urine creatinine were measured by isotope dilution LC-MS/MS as previously described58, and creatinine clearance was calculated. Plasma blood urea nitrogen (BUN) levels and urine albumin were measured by ELISA (Elabscience Biotechnology Co., Bethesda, MD, and GenWay Biotech, Inc, San Diego, CA, respectively).
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay
Detection of apoptotic cells in renal tissue slides was performed using the Apoptag® Plus Peroxidase In Situ Apoptosis Kit (MP Biomedicals, Santa Ana, CA), following manufacturer’s directions. TUNEL-positive cells in tissue sections were counted in 10 microscopic fields (400 × 400 μm, 200X magnification) of renal cortex and outer medulla. TUNEL+ counts are reported as average of the counts in the 10 fields per kidney region.
Statistical analysis
All data are expressed as mean ± SEM. Differences between genotypes and treatments were analyzed by two-way analysis of variance with a Tukey’s post hoc test. A P value of less than 0.05 was considered statistically significant. All statistical analyses were conducting using GraphPad Prism 6 (GraphPad Software, La Jolla, CA).
Additional Information
How to cite this article: De Miguel, C. et al. Endothelin receptor-specific control of endoplasmic reticulum stress and apoptosis in the kidney. Sci. Rep. 7, 43152; doi: 10.1038/srep43152 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Lankhorst, S., Kappers, M. H., van Esch, J. H., Danser, A. H. & van den Meiracker, A. H. Mechanism of hypertension and proteinuria during angiogenesis inhibition: evolving role of endothelin-1. J Hypertens 31, 444–454 (2013).
Orisio, S. et al. Renal endothelin gene expression is increased in remnant kidney and correlates with disease progression. Kidney Int 43, 354–358 (1993).
Kohan, D., Inscho, E. W., Wesson, D. & Pollock, D. M. Physiology of endothelin and the kidney. Compr Physiol 1, 883–919 (2011).
Taniguchi, M. & Yoshida, H. Endoplasmic reticulum stress in kidney function and disease. Curr Opin Nephrol Hypertens 24, 345–350 (2015).
Schönthal, A. H. Endoplasmic reticulum stress: its role in disease and novel prospects for therapy. Scientifica 2012 (2012).
Kawakami, T. et al. Endoplasmic reticulum stress induces autophagy in renal proximal tubular cells. Nephrol Dial Transplant 24, 2665–2672, doi: 10.1093/ndt/gfp215 (2009).
Pallet, N. et al. Cyclosporine-induced endoplasmic reticulum stress triggers tubular phenotypic changes and death. American journal of transplantation: official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 8, 2283–2296, doi: 10.1111/j.1600-6143.2008.02396.x (2008).
Peyrou, M., Hanna, P. E. & Cribb, A. E. Cisplatin, gentamicin, and p-aminophenol induce markers of endoplasmic reticulum stress in the rat kidneys. Toxicol Sci 99, 346–353, doi: 10.1093/toxsci/kfm152 (2007).
Yang, D., Yang, D., Jia, R. & Tan, J. Na+/Ca2+ exchange inhibitor, KB-R7943, attenuates contrast-induced acute kidney injury. J Nephrol 26, 877–885 (2013).
Yang, Y., Yang, D., Yang, D., Jia, R. & Ding, G. Role of reactive oxygen species-mediated endoplasmic reticulum stress in contrast-induced renal tubular cell apoptosis. Nephron Exp Nephrol 128, 30–36 (2014).
Noh, M. R., Kim, J. I., Han, S. J., Lee, T. J. & Park, K. M. C/EBP homologous protein (CHOP) gene deficiency attenuates renal ischemia/reperfusion injury in mice. Biochim Biophys Acta 1852, 1895–1901 (2015).
Zager, R. A., Johnson, A. C., Andress, D. & Becker, K. Progressive endothelin-1 gene activation initiates chronic/end-stage renal disease following experimental ischemic/reperfusion injury. Kidney Int 84, 703–712 (2013).
Yang, C. C., Yao, C. A., Yang, J. C. & Chien, C. T. Sialic acid rescues repurified lipopolysaccharide-induced acute renal failure via inhibiting TLR4/PKC/gp91-mediated endoplasmic reticulum stress, apoptosis, autophagy, and pyroptosis signaling. Toxicol Sci 141, 155–165 (2014).
Jesmin, S. et al. Effects of protease activated receptor (PAR)2 blocking peptide on endothelin-1 levels in kidney tissues in endotoxemic rat mode. Life Sci 102, 127–133 (2014).
Sagar, S. K., Zhang, C., Guo, Q. & Yi, R. Role of expression of endothelin-1 and angiotensin-II and hypoxia-inducible factor-1α in the kidney tissues of patients with diabetic nephropathy. Saudi J Kidney Dis Transpl 24, 959–964 (2013).
Liu, J. et al. Receptor for advanced glycation end-products promotes premature senescence of proximal tubular epithelial cells via activation of endoplasmic reticulum stress-dependent p21 signaling. Cell Signal 26, 110–121 (2014).
Yeager, M. et al. Endothelin-1, the unforlded protein response, and persistent inflammation. Am J Respir Cell Mol Biol 46, 14–22 (2012).
Jain, A., Olovsson, M., Burton, G. J. & Yung, H. W. Endothelin-1 induces endoplasmic reticulum stress by activating the PLC-IP(3) pathway: implications for placental pathophysiology in preeclampsia. Am J Pathol 180, 2309–2320 (2012).
Padilla, J. & Jenkins, N. T. Induction of endoplasmic reticulum stress impairs insulin-stimulated vasomotor relaxation in rat aortic rings: role of endothelin-1. J Physiol Pharmacol 64, 557–564 (2013).
Metcalfe, W. How does early chronic kidney disease progress? A Background Paper prepared for the UK Consensus Conference on Early Chronic Kidney Disease. Nephrol Dial Transplant 22, ix26–ix30 (2007).
Cattaruzza, M., Dimigen, C., Ehrenreich, H. & Hecker, M. Stretch-induced endothelin B receptor mediated apoptosis invascular smooth muscle cells. FASEB J 14, 991–998 (2000).
Hocher, B. et al. Apoptosis in kidneys of endothelin-1 transgenic mice. J Cardiovascr Pharm 31, S554–S556 (1998).
Wu-Wong, J., Chiou, W. J., Dickinson, R. & Opgenorth, T. J. Endothelin attenuates apoptosis in human smooth muscle cells. Biochem J 328, 733–737 (1997).
Dong, F. et al. Endothelin-1 enhances oxidative stress, cell proliferation and reduces apoptosis in human umbilical vein endothelial cells: role of ETB receptor, NADPH oxidase and caveolin-1. Br J Pharmacol 145, 323–333 (2005).
Shichiri, M., Sedivy, J. M., Marumo, F. & Hirata, Y. Endothelin-1 is a potent survival factor for c-Myc-dependent apoptosis. Mol Endocrinol 12 172–180 (1998).
Zinszner, H. et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes & Development 12, 982–995 (1998).
Lhotak, S. et al. ER stress contributes to renal proximal tubule injury by increasing SREBP-2-mediated lipid accumulation and apoptotic cell death. Am J Physiol Renal Physiol 303, F266–278, doi: 10.1152/ajprenal.00482.2011 (2012).
Hodeify, R. et al. Gender differences control the susceptibility to ER stress-induced acute kidney injury. Am J Physiol Renal Physiol 304, F875–F882 (2013).
Samali, A., FitzGerald, U., Deegan, S. & Gupta, S. Methods for Monitoring Endoplasmic Reticulum Stress and the Unfolded Protein Response. International Journal of Cell Biology 2010 (2010).
Gariepy, C. E. et al. Transgenic expression of the endothelin-B receptor prevents congenital intestinal aganglionosis in a rat model of Hirschsprung disease. Journal of Clinical Investigation 102, 1092 (1998).
Simonson, M. & Ismail-Beigi, F. Endothelin-1 increases collagen accumulation in renal mesangial cells by stimulating a chemokine and cytokine autocrine signaling loop. J Biol Chem 286, 11003–11008 (2011).
Sorokin, A. & Kohan, D. E. Physiology and pathology of endothelin-1 in renal mesangium. Am J Physiol Renal Physiol 285 (2003).
Saleh, M. A. et al. Endothelin receptor A-specific stimulation of glomerular inflammation and injury in a streptozotocin-induced rat model of diabetes. Diabetologia 54, 979–988, doi: 10.1007/s00125-010-2021-4 (2011).
Chiang, C. K. et al. Endoplasmic reticulum stress implicated in the development of renal fibrosis. Mol Med 17, 1295–1305 (2011).
Zhang, K. & Kaufman, R. J. From endoplasmic-reticulum stress to the inflammatory response. Nature 454, 455–462 (2008).
Sedeek, M. et al. Role of reactive oxygen species in endothelin-induced hypertension. Hypertension 42, 806–810 (2003).
Li, G., Scull, C., Ozcan, L. & Tabas, I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol 191, 1113–1125 (2010).
Wu-Wong, J. R. & Opgenorth, T. J. Specific Inhibition of Glycosylation by Tunicamycin Affects Endothelin Receptors in Cultured Swiss 3T3 Fibroblasts. Endothelium 1, 153–160, doi: 10.3109/10623329309102691 (1993).
Gellai, M., DeWolf, R., Pullen, M. & Nambi, P. Distribution and functional role of renal ET receptor subtypes in normotensive and hypertensive rats. Kidney Int 46, 1287–1294 (1994).
Taylor, T. A., Gariepy, C. E., Pollock, D. M. & Pollock, J. S. Unique endothelin receptor binding in kidneys of ETB receptor deficient rats. Am J Physiol Regul Integr Comp Physiol 284, R674–681 (2003).
Hirata, Y. et al. Endothelin receptor subtype B mediates synthesis of nitric oxide by cultured bovine endothelial cells. J Clin Invest 91, 1367–1373 (1993).
Plato, C. F., Pollock, D. M. & Garvin, J. L. Endothelin inhibits thick ascending limb chloride flux via ETB receptor-mediated NO release. Am J Physiol Renal Physiol 279, F326–F333 (2000).
Kelsen, S., Hall, J. E. & Chade, A. R. Endothelin-A receptor blockade slows the progression of renal injury in experimental renovascular disease. Am J Physiol Renal Physiol 301, F218–225 (2011).
Hocher, B. et al. ETA receptor blockade induces tubular cell proliferation and cyst growth in rats with polycystic kidney disease. J Am Soc Nephrol 14, 367–376 (2003).
Chang, M. Y. P. E., El Nahas M., Haylor J. L. & Ong A. C. Endothelin B receptor blockade accelerates disease progression in a murine model of autosomal dominant polycystic kidney disease. JASN 18, 560–569 (2007).
Yoo, K. et al. Endothelin A receptor blockade influences apoptosis and cellular proliferation in the developing rat kidney. J Korean Med Sci 24, 138–145 (2009).
Ogata, Y. et al. Antiapoptotic effect of endothelin-1 in rat cardiomyocytes in vitro . Hypertension 41, 1156–1163 (2003).
Shichiri, M., Yokokura, M., Marumo, F. & Hirata, Y. Endothelin-1 inhibits apoptosis of vascular smooth muscle cells induced by nitric oxide and serum deprivation via MAP kinase pathway. Arterioscler Thromb Vasc Biol 20, 989–997 (2000).
Sirén, A. et al. Endothelin B receptor deficiency augments neuronal damage upon exposure to hypoxia-ischemia in vivo . Brain Res 945, 144–149 (2002).
Shichiri, M., Kato, H., Marumo, F. & Hirata, Y. Endothelin-1 as an autocrine/paracrine apoptosis survival factor for endothelial cells. Hypertension 30, 1198–1203 (1997).
Lahav, R., Suvà, M. L., Rimoldi, D., Patterson, P. H. & Stamenkovic, I. Endothelin receptor B inhibition triggers apoptosis and enhances angiogenesis in melanomas. Cancer Res 64, 8945–8953 (2004).
Mencarelli, M. et al. Endothelin receptor A expression in human inflammatory cells. Regul Pept 158, 1–5 (2009).
Guruli, G. et al. Function and survival of dendritic cells depend on endothelin-1 and endothelin receptor autocrine loops. Blood 104, 2107–2115 (2004).
Denny, M. F. et al. Accelerated macrophage apoptosis induces autoantibody formation and organ damage in systemic lupus erythematosus. J Immunol 176, 2095–2104 (2006).
Liu, G. et al. Apoptosis induced by endoplasmic reticulum stress involved in diabetic kidney disease. Biochemical and biophysical research communications 370, 651–656 (2008).
Lim, M. P., Devi, L. A. & Rozenfeld, R. Cannabidiol causes activated hepatic stellate cell death through a mechanism of endoplasmic reticulum stress-induced apoptosis. Cell death & disease 2, e170, doi: 10.1038/cddis.2011.52 (2011).
Lipson, K. L., Ghosh, R. & Urano, F. The role of IRE1alpha in the degradation of insulin mRNA in pancreatic beta-cells. PloS one 3, e1648 (2008).
Takahashi, N. et al. Tandem mass spectrometry measurements of creatinine in mouse plasma and urine for determining glomerular filtration rate. Kidney Int 71, 266–271, doi: 10.1038/sj.ki.5002033 (2007).
Acknowledgements
These studies were funded in part by AHA 11POST7690066 and NIH T32 DK007545 to CDM, and P01 HL95499 to DMP and JSP. We thank the UAB-UCSD O’Brien Center (NIH DK079337) for technical support in measuring urine and plasma creatinine.
Author information
Authors and Affiliations
Contributions
C.D.M. performed experiments, prepared figures and wrote and edited the manuscript. W.C.H. and J.L.H. performed experiments and approved the manuscript. D.M.P., P.K.C. and J.S.P. prepared, reviewed, and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
De Miguel, C., Hamrick, W., Hobbs, J. et al. Endothelin receptor-specific control of endoplasmic reticulum stress and apoptosis in the kidney. Sci Rep 7, 43152 (2017). https://doi.org/10.1038/srep43152
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep43152
This article is cited by
-
Endothelin 1-induced retinal ganglion cell death is largely mediated by JUN activation
Cell Death & Disease (2020)
-
Review of Silicon Recovery and Purification from Saw Silicon Powder
JOM (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
