
Abstract
The origin and domestication of cultivated barley have long been under debate. A population-based resequencing and phylogenetic analysis of the single copy of RPB2 gene was used to address barley domestication, to explore genetic differentiation of barley populations on the worldwide scale, and to understand gene-pool exchanges during the spread and subsequent development of barley cultivation. Our results revealed significant genetic differentiation among three geographically distinct wild barley populations. Differences in haplotype composition among populations from different geographical regions revealed that modern cultivated barley originated from two major wild barley populations: one from the Near East Fertile Crescent and the other from the Tibetan Plateau, supporting polyphyletic origin of cultivated barley. The results of haplotype frequencies supported multiple domestications coupled with widespread introgression events that generated genetic admixture between divergent barley gene pools. Our results not only provide important insight into the domestication and evolution of cultivated barley, but also enhance our understanding of introgression and distinct selection pressures in different environments on shaping the genetic diversity of worldwide barley populations, thus further facilitating the effective use of the wild barley germplasm.
Similar content being viewed by others

Introduction
Barley is one of the oldest, most widely distributed, extensively cultivated, and economically important crops. Cultivated barley (Hordeum vulgare L.) is the domesticated descendants of wild barley (Hordeum spontaneum L.)1. Domestication is the outcome of a selection process that led to increased adaptation to cultivation or rearing and utilization by humans2. Previous studies have demonstrated much less variation in cultivated barley in relative to wild barley3,4,5,6,7, suggesting that cultivated barley originated from small initial wild populations8. However, sites of barley domestication events remain under debate. The oldest archaeological remains of barley grains were found at various human Neolithic sites in the Fertile Crescent and traced back to around 8500 calibrated years (cal.) B.C.1,9,10,11. The wider distribution wild barley in the Near East Fertile Crescent as well as historic and molecular studies commonly supported that the Near East Fertile Crescent is both a major original center of wild barley and a domestication center of its cultivated form3,9,12,13,14.
However, since the multiple domestication centers of barley were postulated as early as 192615, the original center of barley cultivation has been widely debated. In addition to the primary habitats of the Fertile Crescent, the natural distribution of H. spontaneum in several geographically distinct locations has challenged the prevalent monocentric theory of the origin of barley16,17. Multiple domestications imply independent origins of many agronomically important mutations18,19. Studies on row-type of barley demonstrated that six-rowed phenotype originated repeatedly, at different times and in different regions independently, through mutations of Vrs120. Distinct genetic loci determining traits-brittle rachis were found in Eastern and Western barleys18,21. The concept of polyphyletic domestication of cultivated barley was also bolstered by numerous genetic studies22,23,24,25. Recent resequencing data from multiple loci, for instance, proposed that barley has been domesticated at least twice in two locations, within the Fertile Crescent and at location 1,500–3,000 km farther East22.
The role of wild barley from the Tibetan Plateau in the process of the origin and evolution of cultivated barley has attracted increasing attention26,27,28,29,30,31,32. Morphological, archaeological, cytogenetic and isozyme data revealed that wild barley on the Qinghai-Tibet Plateau is different from that in the Fertile Crescent33. Diversity array technology (DArT) data and population-based phylogenetic analyses indicated that the Tibetan Plateau and its vicinity is one of the domestication centers of cultivated barley32,33. Recent transcriptome profiling and population-based genetic diversity analysis also provided strong evidence that barley domestication may have occurred independently in geographically distinct regions34,35. However, in comparison to the abundant works on the Fertile Crescent and Central Asia, an Eastern center of origin and domestication of barley has long been underestimated32. Additional evidence is still needed to shed further light on cultivated barley domestication, in particular, the position of Qinghai-Tibet Plateau wild barley in origin and domestication events.
The varied evolutionary histories of wild barleys and widely dispersed landraces have generated diverse ecotypes, due to natural or human selection, resulting in a wide range of phenotypic/genotypic characteristics36,37,38. Over recent years molecular population genetics has been widely used to investigate genetic diversity within and among barley populations, and to trace the population structure and domestication events22,36,39,40,41,42. However, few investigations have been undertaken to examine genetic differentiation of barley on a worldwide scale, and, particularly, in relation to understanding geographic expansion and introgression.
Resequencing candidate genes can identify all mutations in a particular gene, thus allowing population-based analyses of genetic variation43. Recent advances in the phylogenetic and domestication history analysis with specific resequencing on multiple loci have been widely available in many crops25,36,44,45,46. However, not all genes reflect the history of a crop accurately. Although the majority of the genes in the genome will represent the true history of a domesticated lineage, domestication genes might falsely indicate incorrect origin47. Single copy nuclear genes hold a great potential to improve the robustness of phylogenetic reconstruction at all taxonomic levels, especially when universal markers such as cpDNA and/or nrDNA, are unable to generate strong phylogenetic hypotheses48. Single-copy nuclear genes are advantageous for studying the origin and phylogeny of species because of their high content of functional information and a modest rate of evolutionary change48,49. In this work, population-based resequencing and phylogenetic analysis of the second largest subunit of RNA polymerase II (RPB2) were performed. Nuclear RNA polymerases in eukaryotes have three distinct classes, which are frequently referred to as RNA polymerase I, II, and III. Each enzyme is composed of two large (>100 kDa) and several smaller subunits, each of which is typically encoded by a unique single-copy gene50. RPB2 encodes the second-largest subunit of nuclear RNA polymerase II, which forms a part of the catalytic core that is believed to function in nucleotide binding and RNA chain elongation, and is responsible for the transcription of protein-encoding genes51,52. The only complete RPB2 sequence in plants has been identified in Arabidopsis thaliana, which is 3,564 bp in length with 24 introns53. This gene is found in all eukaryotes, and large regions are highly conserved50. It has been demonstrated that RPB2 is encoded by a single gene in many organisms, including H. vulgare52. A high level of polymorphisms present in this gene indicated that RPB2 is an excellent tool in investigating molecular evolution and phylogenetic relationships54,55,56.
Understanding the origin of crops is important for exploiting elite genetic resources, and in helping to illuminate the history of domestication that would explain further the origin and development of modern cultivation and agronomy2. However, as mentioned above, the pattern of barley domestication is still controversial, information on geographically based genetic differentiation of barley populations on the worldwide scale is poorly documented, and how gene pool exchanges during spread and subsequent development of barley cultivation in the world remains to be explored. We used the RPB2 gene to analyze the genetic variation among geographically distinct barley populations distributed worldwide. The objectives of our study were (i) to investigate genetic differentiation among wild barleys from the Near East Fertile Crescent and Tibetan Plateau populations, and between wild barley and cultivated barley sourced from different geographical regions; (ii) to address contentious points of barley domestication; and (iii) to examine introgression among worldwide barley populations.
Results
Haplotype analysis in barley populations
Of the 212 genotypes screened, 21 distinguishable haplotypes were identified. Haplotype compositions and frequencies in three wild barley populations and six cultivated barley populations were summarized in Table 1. A total of 21 haplotypes were identified in the 88 wild barley accessions, of which 18 haplotypes were identified in the Southwest Asian, 5 in the Central Asian and 4 in the Tibetan wild barley populations. Eighteen out of the 21 haplotypes were population specific: 15 specific to the Southwest Asian, 2 specific to the Central Asian and one specific to the Tibetan wild barley population. Only 6 haplotypes were identified in 124 domesticated lines, all 6 were present in the East Asian cultivated barley population, 5 and 4 in the Mediterranean and European cultivated barley population, respectively, and 3 in the remainder of the domesticated populations. However, no cultivated barley population specific haplotype was found. Haplotypes are shown in Supplementary Fig. S1. With the exception of the singleton polymorphisms (those occurring only once in the sample), 10 haplotype-specific SNPs were detected across 8 population-specific haplotypes. Of these, 8 SNPs were unique to the Southwest Asian wild population, and 2 each were unique to Central Asian and Tibetan wild barley.
The haplotype frequencies present in all sampled accessions ranged from 0.005 to 0.325. Among all the haplotypes across the 212 accessions, 4 major haplotypes were detected. More than half of the accessions screened (119 of 212) have either haplotype Hap 1 or Hap 2, with Hap10 observed in 25 accessions (11.8%), and the Hap12 observed in 24 accessions (11.3%). The frequency of the other 17 haplotypes was low, ranging from 0.5% to 5.7%. RPB2 haplotype frequencies differed markedly in different geographical populations. This was particularly evident for the haplotype Hap1, which was most frequent in Tibetan wild barleys and East Asian cultivars (0.65 and 0.508, respectively), but absent in North American and Australian cultivated barleys, and rarely present in the remaining five barley populations. Also noticeable was absence of the Hap10 in all cultivated populations, which was rare in the Tibetan wild barley population (0.05), but the most frequent in the Central Asian and Southwest Asian wild barley populations (0.60 and 0.25, respectively). These rare haplotypes were confined to specific geographical regions. i.e., of the 14 haplotypes that were present in <2% of the accessions sampled, 12 haplotypes were unique to the Southwest Asian wild barley population and 2 haplotypes to the Central Asian wild barley population (Table 1; Fig. 1).

Geographic distribution of wild barley populations and cultivated barley populations, and RPB2 haplotype frequencies among nine geographic regions.
RPB2 haplotype frequencies were displayed in pie diagrams and the proportion was given in percentage each with different color. The haplotype was calculated with DnaSP version 5.080. The haplotype diagrams in Fig. 1 where generated by using SPSS 20.0 (IBM Corp., Armonk, NY, USA) and manually added to the map using Adobe Photoshop 9.0 (Adobe Systems Inc., San Jose, CA, USA). The original map was acquired from Google Maps (Map data: Google, NASA, TerraMetrics).
Genetic diversity analysis and neutrality test
As shown in Table 2, the highest number of haplotypes (H = 21) and highest number of segregating sites (S = 21), as well as the greatest per-site nucleotide diversity (θ = 0.00558 ± 0.00181), haplotype diversity (Hd = 0.747) and nucleotide diversity (π = 0.00307) were observed in wild barley, while 13.5% haplotype diversity (Hd) and 18.2% nucleotide diversity (π) reduction were found in cultivated barley. Both Tajima’s D, and Fu and Li’s statistics were positive for cultivated barley, but negative for wild barley. Fu and Li’s values were significant (P < 0.05) for wild barley. However, for cultivated barley, Tajima and Fu and Li’s neutrality tests did not significantly depart from neutrality.
To reveal domestication pressures acting on geographically distinct barley populations and the genetic differentiation among them, genetic analysis and the neutrality test in different populations were further performed (Table 3). The highest number of haplotypes (H = 18), highest haplotype diversity (Hd = 0.785), and greatest per-site nucleotide diversity (θ = 0.00575 ± 0.00203) were observed in the Southwest Asian wild barley population among the three wild barley populations. The nucleotide diversity as measured by π was 0.00342, ranging from 0.00098 in the Central Asian wild barley population to 0.00352 in the Mediterranean coast landrace. Both Tajima and Fu and Li’s neutrality tests were not significant (P > 0.05) in all six cultivated populations as well as in the Tibetan and Central Asian wild barley populations. Positive values for both tests were obtained from the North American and European cultivated populations, as well as from Tibetan wild barley population. In contrast, both negative values were obtained from the East Asian, South American, Mediterranean Coast and Australian cultivated populations. However, Southwest Asian wild barley population showed significant negative Fu and Li’s D and F values (P < 0.05) (−2.52062 and −2.68559, respectively).
Sequence polymorphism analysis
The amplified RPB2 fragments ranged from 745 bp to 858 bp in size. Its structure was further identified according to the published sequence of H. vulgare cDNA (GenBank accession number AF020839) in NCBI (http://www.ncbi.nlm.nih.gov/) (Supplementary Fig. S2). The example of amplified pattern of RPB2 is shown in Fig. 2. Among three wild barley populations, amplicons with size of ~850 bp were detected in 95% of Central Asian wild barley accessions and 71% of Southwest Asian wild barley accessions, but in only 10% accessions of Tibetan wild barley.

Example of an amplified pattern of the RPB2 gene from three wild barley populations and sampled cultivated barleys.
Wb-S (Wild barley of Southwest Asia), Wb-C (Wild barley of Central Asia), Wb-T (Wild barley of Tibet) and Cb (sampled cultivated barley).
Multiple sequence alignments showed that a major of 105-bp deletion was clearly observed in the Tibetan wild barley and most cultivated accessions (108 of 124 accessions) (Fig. 3). However, the deletion in this region was rarely occurred in the Southwest Asian and Central Asian wild barley.

Partial alignment of the amplified sequences of RPB2 from wild and cultivated barleys.
Dashed lines indicate the deletion sequence. The bases with gray color indicate the single-base mutation compared to the same position of other samples. The accessions belongs to Wb-T (Wild barley of Tibet), Wb-C (Wild barley of Central Asia) and Wb-S (Wild barley of Southwest Asia) and Cb (sampled cultivated barley).
Phylogenetic and STRUCTURE analysis
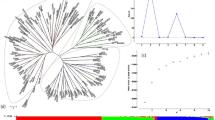
Multi-method phylogenetic analyses generated nearly identical topologies (data not shown). Neighbor-joining tree based on Tajima-Nei distance was shown here. Phylogenetic analysis of wild barley showed a separation of the Tibetan wild barleys (cluster I) from the most of Near East and Central Asian wild barleys (cluster II) (Supplementary Fig. S3). All 212 accessions were divided into two clusters (Fig. 4). The first contained the majority of wild barley accessions (red bar in Fig. 4) and the second cluster contained the majority of cultivated barley accessions (green bar in Fig. 4). However, the most of Tibetan wild barleys (18 of 20 accessions) and some Southwest Asian wild barleys (14 of 48 accessions) were distinct from the wild-dominated cluster, and appeared in the cultivars-dominated cluster.

Phylogenetic tree (neighbor-joining) of 212 total barley accessions based on the RPB2 gene.
Two clusters, one comprised of a majority of wild barley accessions (represented in red bar) and another comprised of a majority of cultivated barley accessions (represented in green bar) are separated. The square stands for wild barley accessions: Tibet (Wb-T, red), Southwest Asia (Wb-S, purple), and Central Asia (Wb-C, orange), respectively; the triangle indicates cultivated barleys: East Asia (Cb-EA, black), North America (Cb-NA, blue), South America (Cb-SA, pink), Mediterranean Coast Areas (Cb-MA, green), Europe (Cb-EU, yellow), and Australia (Cb-AU, brown).
STRUCTURE analysis revealed a clear evolutionary divergence between Near East and Tibetan wild barley (Supplementary Fig. S4). About 90% Tibetan wild barleys (18 of 20) with high membership coefficients of Q ≥ 0.991 were assigned to the population 1 (Q1 in Supplementary Fig. S4C), while 83.3% Southwest Asian wild barley (40 of 48) and 95% Central Asian wild barley accessions (19 of 20) were assigned to the population 2 (Q2 in Supplementary Fig. S4C), with membership coefficients of Q from 0.828 to 0.997. Structure analysis of all 212 barley accessions detected two groups, a wild-dominated group (Q1 in Fig. 5C) consisted mainly of most of wild barley accessions, and an admixed group (Q2 in Fig. 5C), which contained the most of cultivated barley, some Tibetan and Southwest Asian wild barleys. Cluster and structure analysis were also performed for 124 cultivated barley (data not shown): however, no visible subpopulation feather were recognized, which showed an admixed state, shown in Figs 4 and 5. All of the information of the STRUCTURE analysis results and inferred ancestry of individuals were shown in Supplementary Tables S1 and S2.

Analysis of population structure of 212 total barley accessions using STRUCTURE.
(A) Estimated LnP(K) of possible clusters (K) from 1 to 10. (B) ΔK based on rate of change of LnP (K) between successive K values. (C) Population structure based on K = 2. Red represents Subgroup Q1; green, Subgroup Q2. Each part of figure was manually combined using Adobe Photoshop 9.0 (Adobe Systems Inc., San Jose, CA, USA).
Discussion
Genetic differentiation among wild barley populations
Previous studies have provided evidence demonstrating a clear genetic differentiation among wild barley populations from Eastern and Central Asia with those from Near East areas8,18,21,57,58,59,60,61. Significant differentiation in roughly half of the sequenced loci from wild barley occurred between the Oriental and Occidental portion of the species22,59,62. Using resequencing data, Morrell and Clegg22 identified the differences in haplotype frequency at multiple loci between Fertile Crescent and Central Asian wild barley. Fang et al.61 recently found a strong genetic differentiation between the Eastern and Western populations on 2H and 5H. Previous morphological, distributional, archaeological, cytogenetic, and isozyme studies have also demonstrated that Tibetan wild barley was different from the Fertile Crescent samples33, which was also supported by the genome-wide DArT data32, transcriptome profiling34, and population-based genetic diversity analysis35. The current results showed significant genetic differentiation among wild barley populations. The distinct haplotype composition and obvious sequence variation were detected among Tibetan wild barley, Central Asian wild barley, and Southwest Asian wild barley (Table 1; Figs 1, 2 and 3; Supplementary Figs S1 and S2). Our phylogenetic analysis and population structure analysis also showed a certain degree of separation among Tibetan, Southwest Asian, and Central Asian wild barleys (Supplementary Figs S3 and S4). Our results provided further evidence to support multiple origination hypothesis of cultivated barley21,22,32, favoring that the wild barley domestication occurred in multiple geographically distinct regions.
Tibet is a domestication center of cultivated barley
Since the discovery of H. agriocrithon E. Åberg, a close wild relative of barley, and of numerous H. spontaneum on the Qinghai-Tibet Plateau, the position of wild barley from the Tibetan Plateau in the process of origin and domestication of cultivated barley has received more attention and debate33. Extensive studies have reported that Tibetan wild barley was clearly different from other areas, and suggested that the Tibetan Plateau and its vicinity are the center of origin for cultivated barley in the Oriental region29,30,31,40, which was also supported by our data here. This was particularly evident for the haplotype Hap1, which was most frequent in the Tibetan wild barleys and East Asian cultivars (0.65 and 0.508, respectively), and haplotype Hap2 unique to Tibetan wild barley, which was also present in the most accessions of East Asian cultivated barleys (Table 1). Furthermore, multiple sequence alignments revealed a 105-bp deletion occurred in most accessions of Tibetan wild barleys, which also occurred in up to 95% of East Asian cultivars (Figs 2 and 3). Consequently, our results suggested that the East Asian cultivated barley might be evolved from the Tibetan wild barley, which is consistent with the report that barley landraces reflect a pattern of over shared ancestry with geographically proximate wild barley populations63. The present data thus provided further evidence to support the hypothesis that that Tibetan wild barley was the ancestor of Oriental domesticated barley33,64.
Our results not only merely confirmed that Tibetan wild barley contributed largely to East Asian cultivars as demonstrated above, but also revealed that these wild germplasms have important contribution to the cultivated barley gene pools outside the Oriental region. The haplotype analysis showed that the cultivars outside East Asia shared the same haplotypes with the wild barley from the Tibet (Table 1; Fig. 1). Sequence comparisons, phylogenetic and population structure analyses also revealed a close relationship between worldwide domesticated barley and the Tibetan wild barley (Figs 2, 3, 4 and 5). Our data confirmed that Tibetan Plateau is one of the centers of domestication of cultivated barley32,34,35.
Multiple domestication and introgression of modern worldwide barleys
Hypotheses of the origin of barley have indicated that if the wild progenitor showed significant difference in allele frequencies among geographical regions, allelic composition is especially likely to be informative as to the number and locations of origin of domesticates22. For wild barley, the region with the highest level of genetic diversity is also most likely center of origin for the cultivated one42. In our study, highest number of haplotypes, greatest haplotype diversity and per-site nucleotide diversity were observed in the Southwest Asian wild barley population, which thus further confirmed that the Near East Fertile Crescent is a primary origin center of cultivated barley (Table 3). Additionally, the distinct haplotypes were detected not only in Southwest Asian wild barley, but also in Tibetan and Central Asian wild barleys (Table 1; Fig. 1). A great difference among distinct wild barleys, and a close relationship between these wild barleys and domesticated barley were revealed in our study, suggesting that Southwest Asian, Central Asian, and Tibetan wild barley are the ancestors of cultivars. Our results thus supported multiple origins of cultivated barley22,32.
In addition, the haplotypes analysis revealed that a significant proportion of the genetic composition of Eastern and Western wild barley has spread cultivars in other regions of the world. For example, haplotypes unique to Eastern wild barley (from Tibetan wild barley population) were also present in Occidental landraces, and haplotypes private to Western wild barley (from Southwest Asian wild barley population) were also found in Oriental landraces (Table 1; Fig. 1). As we observed, previous studies also reported that a significant proportion of Western genetic composition appeared in Indian and East Asian barleys, and the Eastern alleles were also found in Occidental landraces25,32,65. It was suggested that Central Asia is the sole route for wild barley migration between the Near East and the Tibetan Plateau32, as inferred in our haplotypes analysis; Hap1, Hap10 and Hap12 were shared among three wild barley populations and are most frequent in the Tibetan or Southwest Asian wild barleys, while rare in Central Asian wild barley population (Table 1; Fig. 1).
Consequently, our study provides new perspective on barley domestication and worldwide cultivation. We suggested that worldwide introgression has occurred following multiple domestication events, and, in this process Near East and Tibetan wild barleys have contributed to the modern cultivated barley gene pool.
Our scenario on barley origin and domestication may also offer an alternative explanation on why high genetic diversity and numbers of private haplotypes were present in Near East wild barley (Table 1; Table 3), and why specific haplotypes in Tibetan wild barley seem more widely present in cultivars at some locations and a close relationship between Tibetan wild and cultivated barley, as shown in previous reports32,33,35, as well in this study (Table 1; Fig. 1). Firstly, Near East Hordeum spontaneum is widely distributed as wild populations but largely isolated from cultivated barley1,3,9. However, wild barley in Tibet always coexists as a weed with cultivated barley and other field crops27, allowing gene flow to occur more easily between the two32. A long period of gene flow may have led to subsequent transfer of introgressed haplotypes to cultivars in other regions due to human activities such as germplasm exchange, introduction and hybridization35.
Natural variation in the barley population
Domestication is the outcome of a selection process that led to increased adaptation to cultivation and utilization by humans2. Gene pools undergoing domestication experienced dramatic changes in allele frequencies due to genetic bottleneck and drift or selection, and some allelic combinations may be lost37,38,66. As expected, in this study, among the 21 haplotypes of RPB2 sequence found in 212 barley accessions, only eight were present in the domesticated lines (Table 2), which agreed well with previous reports33,35,67, indicating that domesticated lines have lost most alleles in wild types7,33,68,69. About 18.2% nucleotide diversity, 13.5% haplotype diversity and two-fold of per-site nucleotide diversity reduction in cultivated barley, which is consistent with the studies such as Fu43 and Morrell et al.25, suggested that barley landraces might have suffered a population bottleneck during domestication and resulted in a reduction in genetic diversity68. Genetic bottleneck due to domestication and breeding is the major determinant of polymorphism loss in the domesticated lines sampled67. This loss is evident in a shift toward more positive values of Tajima’s D in the domesticated relative to wild populations25,35. Similarly, in our study, positive values of Tajima’s D and Fu, and Li’s were found in cultivated barley, while negative values were found in wild barley (Table 2). This is consistent with previous studies70,71 and supports that genetic bottleneck tends to result in a loss of rare variants72. RPB2 showed significant negative values of Fu and Li’s D and F when all wild barley were considered (Table 2); this could potentially indicate a deviation from neutrality, possibly due to positive selection67. However, both Tajima’s D, and Fu and Li’s values in all cultivated barley were insignificant positive, which may have resulted from balancing selection or bottleneck effect.
It was notable that the genetic diversity in some domesticated barley populations was higher than that in wild barley populations, which is consistent with previous observations of the same gene in Vitis vinifera56, but in contrast with what we have demonstrated above that the gene pool of whole cultivated barley suffered a reduction in genetic diversity. We suggest that there are two possible explanations. Firstly, this might be caused by the nature of the RPB2 gene, as it encodes the second largest subunit of nuclear RNA polymerase II, and is responsible for the transcription of protein encoding genes, which are very important for various aspects of plant life54. The different barley populations are from diverse environments, which could increase selection pressure on RPB2. The second explanation is the higher genetic variability and the higher substitution rate of RPB2 in the domesticated barley as suggested by Zecca and Grassi56, can be viewed as a consequence of natural conditions, human selection, and germplasm exchange and breeding. Tajima’s D, and Fu and Li’s values in cultivated populations vary from positive to negative, indicating that distinct geographical and environmental barley population may be subjected to different selective pressure (Table 3). Balancing selection or bottleneck may act upon North American and European cultivated barley populations where rare-allele advantage resulted in an accumulating allelic frequency up to an intermediate level that may have caused a positive value of Tajima’s D, as suggested by Chung et al.73. However, purifying selection might act on the remaining domesticated barley populations, reflecting a negative statistical values in these regions68. In this study, Tajima’s D, and Fu and Li’s neutrality tests revealed no evidence of natural selection for Tibetan wild barley population, but under purifying selection as revealed by a high statistic positive value. This insignificant result may be attributed to the low polymorphism observed, which weakens the neutrality test. This result agrees with previous reports on CPsHSP-2 in Machilus kusano73. Obviously, deviation from neutrality with Fu and Li’s values was significant (P < 0.05) for Southwest Asian wild barley population, which resulted from the observed number of rare variants that exceeded the expected number in an equilibrium neutral model and could be interpreted as being a result of a selective sweep or a population expansion73.
In summary, our study provided new insights into the origin and domestication of worldwide cultivated barley. The current results showed a clear genetic differentiation among Tibetan, Southwest Asian and Central Asian wild barleys. Tibetan Plateau is one of the domestication centers of cultivated barley. Our data suggested that multiple domestication followed by extensive introgression among modern worldwide cultivated barley. Moreover, our data showed divergent domestication pressures acting on geographically discontinuous barley populations.
Methods
Plant Materials
A total of 212 barley accessions were used in this study including 88 wild barley (Hordeum spontaneum) accessions from different geographic origins and 124 worldwide cultivated barley (Hordeum vulgare) accessions. The wild barley populations included: 48 wild barley accessions from the Southwest Asia (Israel, Jordan, Ethiopia, Lebanon, Azerbaijan, Syria, Iraq, and Turkey); 20 wild barley accessions from Central Asia (Iran, Afghanistan, Pakistan, and Tajikistan); and 20 wild barley from Qinghai-Tibet Plateau. One hundred and twenty-four accessions of cultivated barley were collected from 18 countries: 61 from Eastern Asia, 8 from South America, 18 form North America, 10 from Mediterranean coast areas, 5 form Australia, and 22 from Europe. Those materials were provided by the USDA (United States Department of Agriculture) and Huazhong Agricultural University barley germplasm collection. Information on accession numbers, and geographical origins of individuals used in this investigation are given in Supplementary Table S3.
DNA extraction, RPB2 gene amplification and sequencing
The seeds were planted in pots with nutrient soil, and maintained in a growth chamber with 14 h of light at 22 °C and 10 h of darkness at 18 °C prior to DNA extraction. Young leaves were collected from 5 to 10 plants of each accession. Total genomic DNA was isolated from freeze-dried leaf tissue following the cetyltrimethylammonium bromide (CTAB) extraction method of Stein et al.74. The quality of DNA was checked using 0.8% agarose gel electrophoresis, and further measured using spectrophotometer. The RPB2 gene sequences were amplified using polymerase chain reaction (PCR) with primer P6F (5′-TGGGGAATGATGTGTCCTGC-3′) and P6FR (5′-CGAACCACACCAACTTCAGTGT-3′)54. PCR amplification was performed in Bio-Rad iCycler thermal cycler (Bio-Rad, USA). Each PCR reaction mixture (40 μl) consisting of 60 ng template DNA, 0.2 μM of each primer, 1.5 mM MgCl2, 0.2 mM of each deoxynucleotide (dATP, dCTP, dGTP, dTTP), 1.5 unit of high-fidelity polymerase ExTaq (TaKaRa, Dalian, China), and distilled deionized water was added to make up the final volume of 40 μl. The PCR was programmed at an initial denaturing of 4 min at 95 °C, followed by 40 cycles of 1 min at 95 °C, 1 min annealing temperatures at 56 °C, 2 min extension at 72 °C and a final extension step at 72 °C for 8 min.
The amplified products were separated by electrophoresis in 1% agarose gels, and the single specific PCR product band was purified by the QIAquick PCR purification kit (Qiagen, Germany) according to the manufacturer′s instruction. DNA was sequenced commercially at the Beijing Tsing Ke BioTech Co., Ltd (Beijing, China). To exclude sequencing errors induced by Taq DNA polymerase during PCR amplification, for each accession, the amplifying and sequencing were repeated three times. The final nucleotide sequence was determined from the sequencing results of both forward and reverse strands, and further data quality were checked using Chromas 2.32 (Technelysium Pty. Ltd.).
Data Analysis
Multiple sequence alignments were performed using ClustalX75. Nucleotide diversity was estimated by Tajima′s π76 and Watterson’s77 statistics. Tests of neutral evolution were performed as described by Tajima78, and Fu and Li79. The above calculations were conducted using the software program DnaSP version 5.080. Each insertion/deletion (indel) was considered as a single mutation event, and all indels were therefore coded as single positions. Identical sequences were grouped into haplotypes (Hap). Phylogenetic analysis was performed with the computer program MEGA 681 using the maximum likelihood (ML) method under the Kimura 2-parameter model, the minimum-evolution (ME) and neighbor-joining (NJ) methods with the model of Tajima-Nei. The confidence of each clade was calculated based on the bootstrap values with 1,000 replications.
The population structure was analyzed using STRUCTURE software (version 2.3.4)82,83. Haplotypes were recoded as unique alleles. Multistep approach (after several trial runs) was applied to infer the genetic structure in our wild, cultivated as well as all barley samples, respectively. The first step of the analysis consisted of estimating K-value (the putative number of genetic groups). Twenty independent runs of K from 1 to 10 were performed, with 100,000 MCMC (Markov Chain Monte Carlo) iterations and a burn-in period of 50,000 replicates under the ‘admixture model’. The most likely K-value was estimated by the log probability of data [LnP(D)] and an ad hoc statistic ΔK based on the rate of change of LnP(D) between successive K values as described by Evanno et al.84. To infer the appropriate number of K, STRUCTURE HARVESTER85 (http://taylor0.biology.ucla.edu/structureHarvester/index.php) was used. In a second step, after the inference of K, the STRUCTURE procedure was repeated with a fixed K and 10 independent runs with 50,000 MCMC iterations and a burn-in period of 25,000. An individual was assigned to a certain cluster if its q value was higher than 0.75.
Additional Information
How to cite this article: Wang, Y. et al. Molecular evidence of RNA polymerase II gene reveals the origin of worldwide cultivated barley. Sci. Rep. 6, 36122; doi: 10.1038/srep36122 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Zohary, D., Hopf, M. & Weiss, E. Domestication of Plants in the Old World: The origin and spread of domesticated plants in Southwest Asia, Europe, and the Mediterranean Basin (Oxford University Press, Oxford, UK, 2012).
Badr, A. & El-Shazly, H. Molecular approaches to origin, ancestry and domestication history of crop plants: barley and clover as examples. J. Genet. Eng. Biotechnol. 10, 1–12 (2012).
Nevo, E., Beiles, A. & Zohary, D. Genetic resources of wild barley in the Near East: structure, evolution and application in breeding. Biol. J. Linn. Soc. 27, 355–380 (1986).
Neale, D. B., Saghai-Maroof, M. A., Allard, R. W., Zhang, Q. & Jorgensen, R. A. Chloroplast DNA diversity in populations of wild and cultivated barley. Genetics. 120, 1105–1110 (1988).
Ellis, R. P. et al. Wild barley: a source of genes for crop improvement in the 21st century? J. Exp. Bot. 51, 9–17 (2000).
Nevo, E. Population genetic structure of wild barley and wheat in the Near East Fertile Crescent: regional and local adaptive patterns in Cereal genomics (eds Gupta, P. K. & Varshney, R. K. ) 135–163 (Kluwer Academic Press, Dordrecht, 2004).
Russell, J. et al. A comparison of sequence-based polymorphism and haplotype content in transcribed and anonymous regions of the barley genome. Genome. 47, 389–398 (2004).
Tanno, K., Taketa, S., Takeda, K. & Komatsuda, T. A DNA marker closely linked to the vrs1 locus (row-type gene) indicates multiple origins of six-rowed cultivated barley (Hordeum vulgare L.). Theor. Appl. Genet. 104, 54–60 (2002).
Harlan, J. R. & Zohary, D. Distribution of wild wheats and barley. Science. 153, 1074–1080 (1966).
Diamond, J. Evolution, consequences and future of plant and animal domestication. Nature. 418, 700–707 (2002).
Willcox, G. The distribution, natural habitats and availability of wild cereals in relation to their domestication in the Near East: multiple events, multiple centres. Veg Hist. Archaeobot. 14, 534–541 (2005).
Badr, A. et al. On the origin and domestication history of barley (Hordeum vulgare). Mol. Biol. Evol. 17, 499–510 (2000).
Nevo, E. Genome evolution of wild cereal diversity and prospects for crop improvement. Plant Genet. Resour. 4, 36–46 (2006).
Mascher, M. et al. Genomic analysis of 6,000-year-old cultivated grain illuminates the domestication history of barley. Nat. Genet. 48, 1089–1093 (2016).
Vavilov, N. I. Studies on the origin of cultivated plants. Bull. Appl. Bot. 1, 1–248 (1926).
Molina-Cano, J. L., Igartua, E., Casas, A. M. & Moralejo, M. New views on the origin of cultivated barley In Barley science: Recent advances from molecular biology to agronomy of yield and quality (eds Slafer, G. A., Molina-Cano, J. L., Savin, R., Araus, J. L. & Romagosa, I. ) 15–29 (Haworth Press, Binghamton, NY, 2002).
Igartua, E., Moralejo, M., Casas, A. M., Torres, L. & Molina-Cano, J. L. Whole-genome analysis with SNPs from BOPA1 shows clearly defined groupings of Western Mediterranean, Ethiopian, and Fertile Crescent barleys. Genet Resour. Crop Evol. 60, 251–264 (2013).
Zohary, D. Monophyletic vs. polyphyletic origin of the crops on which agriculture was founded in the Near East. Genet Resour. Crop Evol. 46, 133–142 (1999).
Sang, T. Genes and mutations underlying domestication transitions in grasses. Plant Physiol. 149, 63–70 (2009).
Komatsuda, T. et al. Six-rowed barley originated from a mutation in a homeodomain-leucine zipper I-class homeobox gene. Proc. Natl. Acad. Sci. USA 104, 1424–1429 (2007).
Azhaguvel, P. & Komatsuda, T. A phylogenetic analysis based on nucleotide sequence of a marker linked to the brittle rachis locus indicates a diphyletic origin of barley. Ann. Bot. 100, 1009–1015 (2007).
Morrell, P. L. & Clegg, M. T. Genetic evidence for a second domestication of barley (Hordeum vulgare) east of the Fertile Crescent. Proc. Natl. Acad. Sci. USA. 104, 3289–3294 (2007).
Saisho, D. & Purugganan, M. D. Molecular phylogeography of domesticated barley traces expansion of agriculture in the Old World. Genetics. 177, 1765–1776 (2007).
Fu, Y. B. & Peterson, G. W. Genetic diversity analysis with 454 pyrosequencing and genomic reduction confirmed the eastern and western division in the cultivated barley gene pool. Plant Genome. 4, 226–237 (2011).
Morrell, P. L., Gonzales, A. M., Meyer, K. K. & Clegg, M. T. Resequencing data indicate a modest effect of domestication on diversity in barley: a cultigen with multiple origins. J. Hered. 105, 253–264 (2013).
Åberg, E. Hordeum agriocrithon, a wild six-rowed barley. Ann. Agric. Coll. Sweden. 6, 159–216 (1938).
Xu, T. W. Origin and evolution of cultivated barley in China. Acta Genet. Sin. 9, 440–446 (1982).
Yang, J. L. & Yen, C. Distribution of weedrace barley in China and the center of origin of cultivated barley. Sver. Utsadesforen. Tidskr. 95, 71–78 (1985).
Zhang, Q. F., Yang, G. P., Dai, X. K. & Sun, J. Z. A comparative analysis of genetic polymorphism in wild and cultivated barley from Tibet using isozyme and ribosomal DNA markers. Genome. 37, 631–638 (1994).
Yin, Y. Q., Ma, D. Q. & Ding, Y. Analysis of genetic diversity of hordein in wild close relatives of barley from Tibet. Theor. Appl. Genet. 107, 837–842 (2003).
Gong, X. et al. Comparative analysis of genetic diversity between Qinghai-Tibetan wild and Chinese landrace barley. Genome. 52, 849–861 (2009).
Dai, F. et al. Tibet is one of the centers of domestication of cultivated barley. Proc. Natl. Acad. Sci. USA. 109, 16969–16973 (2012).
Ren, X. F., Nevo, E., Sun, D. F. & Sun, G. L. Tibet as a potential domestication center of cultivated barley of China. PLoS ONE. 8, e62700 (2013).
Dai, F. et al. Transcriptome profiling reveals mosaic genomic origins of modern cultivated barley. Proc. Natl. Acad. Sci. USA. 111, 13403–13408 (2014).
Wang, Y. G., Ren, X. F., Sun, D. F. & Sun, G. L. Origin of worldwide cultivated barley revealed by NAM-1 gene and grain protein content. Front. Plant Sci. 6, 803 (2015).
Jones, H. et al. Population-based resequencing reveals that the flowering time adaptation of cultivated barley originated east of the Fertile Crescent. Mol. Biol. Evol. 25, 2211–2219 (2008).
Poets, A. M. et al. The effects of both recent and long-term selection and genetic drift are readily evident in North American barley breeding populations. G3-Genes Genom. Genet. 6, 609–622 (2016).
Russell, J. et al. Exome sequencing of geographically diverse barley landraces and wild relatives gives insights into environmental adaptation. Nat. Genet. 48, 1024–1030 (2016).
Baum, B. R., Nevo, E., Johnson, D. A. & Beiles, A. Genetic diversity in wild barley (Hordeum spontaneum C. Koch) in the Near East: a molecular analysis using random amplified polymorphic DNA (RAPD) markers. Genet Res. Crop Evol. 44, 147–157 (1997).
Feng, Z. Y., Liu, X. J., Zhang, Y. Z. & Ling, H. Q. Genetic diversity analysis of Tibetan wild barley using SSR markers. Acta Genet. Sin. 33, 917–928 (2006).
Comadran, J. et al. Patterns of genetic diversity and linkage disequilibrium in a highly structured Hordeum vulgare association-mapping population for the Mediterranean basin. Theor. Appl. Genet. 119, 175–187 (2009).
Wang, A. H., Yu, Z. Y. & Ding, Y. Genetic diversity analysis of wild close relatives of barley from Tibet and the Middle East by ISSR and SSR markers. C.R. Biol. 332, 393–403 (2009).
Fu, Y. B. Population-based resequencing analysis of wild and cultivated barley revealed weak domestication signal of selection and bottleneck in the Rrs2 scald resistance gene region. Genome. 55, 93–104 (2012).
Zhu, Q. H., Zheng, X. M., Luo, J. C., Gaut, B. S. & Ge, S. Multilocus analysis of nucleotide variation of Oryza sativa and its wild relatives: severe bottleneck during domestication of rice. Mol. Biol. Evol. 24, 875–888 (2007).
Haudry, A. et al. Grinding up wheat: a massive loss of nucleotide diversity since domestication. Mol. Biol. Evol. 24, 1506–1517 (2007).
Molina, J. et al. Molecular evidence for a single evolutionary origin of domesticated rice. Proc. Natl. Acad. Sci. USA. 108, 8351–8356 (2011).
Gross, B. L. & Olsen, K. M. Genetic perspectives on crop domestication. Trends Plant Sci. 15, 529–537 (2010).
Sang, T. Utility of low-copy nuclear gene sequences in plant phylogenetics. Crit. Rev. Biochem. Mol. Biol. 37, 121–147 (2002).
Liu, Y. J., Whelen, S. & Hall, B. D. Phylogenetic relationships among ascomycetes: evidence from an RNA polymerse II subunit. Mol. Biol. Evol. 16, 1799–1808 (1999).
Sun, G. L., Pourkheirandish, M. & Komatsuda, T. Molecular evolution and phylogeny of the RPB2 gene in the genus Hordeum. Ann. Bot. 103, 975–983 (2009).
Kolodziej, P. A., Woychik, N., Liao, S. M. & Young, R. A. RNA polymerase II subunit composition, stoichiometry, and phosphorylation. Mol. Cell. Biol. 10, 1915–1920 (1990).
Denton, A. L., McConaughy, B. L. & Hall, B. D. Usefulness of RNA polymerase II coding sequences for estimation of green plant phylogeny. Mol. Biol. Evol. 15, 1082–1085 (1998).
Larkin, R. & Guilfoyle, T. The second largest subunit of RNA polymerase II from Arabidopsis thaliana. Nucleic Acids Res. 21, 1038 (1993).
Sun, G. L., Daley, T. & Ni, Y. Molecular evolution and genome divergence at RPB2 gene of the St and H genome in Elymus species. Plant Mol. Biol. 64, 645–655 (2007).
Liao, J. Q. et al. Molecular phylogeny of RNA polymerase II gene reveals the relationships of tetraploid species with St genome (Triticeae: Poaceae). Biochem. Syst. Ecol. 39, 609–618 (2011).
Zecca, G. & Grassi, F. RPB2 gene reveals a phylodemographic signal in wild and domesticated grapevine (Vitis vinifera). J. Syst. Evol. 51, 205–211 (2013).
Kahler, A. L. & Allard, R. W. Worldwide patterns of genetic variation among four esterase loci in barley (Hordeum vulgare L.). Theor. Appl. Genet. 59, 101–111 (1981).
Zhang, Q. F., Saghai-Maroof, M. A. & Yang, G. P. Ribosomal DNA polymorphisms and the Oriental-Occidental genetic differentiation in cultivated barley. Theor. Appl. Genet. 84, 682–687 (1992).
Morrell, P. L., Lundy, K. E. & Clegg, M. T. Distinct geographic patterns of genetic diversity are maintained in wild barley (Hordeum vulgare ssp. spontaneum) despite migration. Proc. Natl. Acad. Sci. USA. 100, 10812–10817 (2003).
Senthil, N. & Komatsuda, T. Inter-subspecific maps of non-brittle rachis genes btr1/btr2 using occidental, oriental and wild barley lines. Euphytica. 145, 215–220 (2005).
Fang, Z. et al. Two genomic regions contribute disproportionately to geographic differentiation in wild barley. G3-Genes Genom. Genet. 4, 1193–1203 (2014).
Morrell, P. L., Toleno, D. M., Lundy, K. E. & Clegg, M. T. Low levels of linkage disequilibrium in wild barley (Hordeum vulgare ssp. spontaneum) despite high rates of self-fertilization. Proc. Natl. Acad. Sci. USA. 102, 2442–2447 (2005).
Poets, A. M., Fang, Z., Clegg, M. T. & Morrell, P. L. Barley landraces are characterized by geographically heterogeneous genomic origins. Genome Biol. 16, 173 (2015).
Jin, X. L. et al. Genetic variants of HvGlb1 in Tibetan annual wild barley and cultivated barley and their correlation with malt quality. J. Cereal Sci. 53, 59–64 (2011).
Ordon, F., Schiemann, A. & Friedt, W. Assessment of the genetic relatedness of barley accessions (Hordeum vulgare s.l.) resistant to soil-borne mosaic-inducing viruses (BaMMV, BaYMV, BaYMV-2) using RAPDs. Theor. Appl. Genet. 94, 325–330 (1997).
Wang, L. et al. Population structure and linkage disequilibrium in six-rowed barley landraces from the Qinghai-Tibetan plateau. Crop Sci. 54, 2011–2022 (2014).
Kilian, B. et al. Haplotype structure at seven barley genes: relevance to gene pool bottlenecks, phylogeny of ear type and site of barley domestication. Mol. Genet. Genomics. 276, 230–241 (2006).
Xia, Y. S. et al. Single nucleotide polymorphisms in HSP17.8 and their association with agronomic traits in barley. PLoS ONE. 8, e56816 (2013).
Jakob, S. S. et al. Evolutionary history of wild barley (Hordeum vulgare subsp. spontaneum) analyzed using multilocus sequence data and paleodistribution modeling. Genome Biol. Evol. 6, 685–702 (2014).
Wright, S. I. et al. The effects of artificial selection on the maize genome. Science. 308, 1310–1314 (2005).
Caldwell, K. S., Russell, J., Langridge, P. & Powell, W. Extreme population-dependent linkage disequilibrium detected in an inbreeding plant species, Hordeum vulgare. Genetics. 172, 557–567 (2006).
Nei, M., Maruyama, T. & Chakraborty, R. The bottleneck effect and genetic variability in populations. Evolution. 29, 1–10 (1975).
Chung, J. D., Lin, T. P., Cheng, Y. P. & Hwang, S. Y. Geographical variation and differential selection modes of paralogs of chloroplast small heat shock protein genes in Machilus kusanoi (Lauraceae). Bot. Stud. 51, 27–36 (2010).
Stein, N., Herren, G. & Keller, B. A new DNA extraction method for high-throughput marker analysis in a large-genome species such as Triticum aestivum. Plant Breed. 120, 354–356 (2001).
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. & Higgins, D. G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 (1997).
Tajima, F. Evolutionary relationship of DNA sequences in finite populations. Genetics. 105, 437–460 (1983).
Watterson, G. A. On the number of segregating sites in genetical models without recombination. Theor. Popul Biol. 7, 256–276 (1975).
Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 123, 585–595 (1989).
Fu, Y. X. & Li, W. H. Statistical tests of neutrality of mutations. Genetics. 133, 693–709 (1993).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 25, 1451–1452 (2009).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics. 155, 945–959 (2000).
Falush, D., Stephens, M. & Pritchard, J. K. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 164, 1567–1587 (2003).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Earl, D. A. & Vonholdt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Acknowledgements
This project was supported in part by the National Natural Science Foundation of China (Grant No. 31228017), an earmarked fund for China Agriculture Research System (CARS-5) and PhD Candidate Research Innovation Project of Huazhong Agricultural University (Program No. 2014bs07).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: D.S. and G.S. Performed the experiments: Y.W. and X.R. Analyzed the data: Y.W., X.R., D.S. and G.S. Contributed reagents/materials/analysis tools: Y.W., X.R., D.S. and G.S. Wrote the paper: Y.W., X.R., D.S. and G.S.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Wang, Y., Ren, X., Sun, D. et al. Molecular evidence of RNA polymerase II gene reveals the origin of worldwide cultivated barley. Sci Rep 6, 36122 (2016). https://doi.org/10.1038/srep36122
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36122
This article is cited by
-
Genetic and Agro-morphological diversity in global barley (Hordeum vulgare L.) collection at ICARDA
Genetic Resources and Crop Evolution (2021)
-
Fine mapping QSc.VR4, an effective and stable scald resistance locus in barley (Hordeum vulgare L.), to a 0.38-Mb region enriched with LRR-RLK and GLP genes
Theoretical and Applied Genetics (2020)
-
Molecular and phenotypic diversity of ICARDA spring barley (Hordeum vulgare L.) collection
Genetic Resources and Crop Evolution (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
