
Abstract
N-methyl-d-aspartate receptors (NMDARs) play a critical role in excitatory synaptic transmission and plasticity in the central nervous systems. Recent genetics studies in schizophrenia (SCZ) show that SCZ is susceptible to NMDARs and the NMDAR signaling complex. In autism spectrum disorder (ASD), several studies report dysregulation of NMDARs as a risk factor for ASD. To further examine the association between NMDARs and SCZ/ASD development, we conducted a mutation screening study of GRIN2B which encodes NR2B subunit of NMDARs, to identify rare mutations that potentially cause diseases, in SCZ and ASD patients (n = 574 and 152, respectively). This was followed by an association study in a large sample set of SCZ, ASD, and normal healthy controls (n = 4145, 381, and 4432, respectively). We identified five rare missense mutations through the mutation screening of GRIN2B. Although no statistically significant association between any single mutation and SCZ or ASD was found, one of its variant, K1292R, is found only in the patient group. To further examine the association between mutations in GRIN2B and SCZ/ASD development, a larger sample size and functional experiments are needed.
Similar content being viewed by others

Introduction
Schizophrenia (SCZ) is a severe chronic psychiatric disease that is characterized by psychotic symptoms such as hallucinations and delusions. The lifetime risk of SCZ is estimated to be 1%, with subsequent mortality of SCZ patients is 2.5 times higher than in the general population1. Autism spectrum disorder (ASD) is characterized by impaired social interactions and communications and by restricted, repetitive behavior. The prevalence of prototypical ASD is around 25 per 10000, and that of broad ASD is 116 per 100002. The heritability of SCZ and ASD is estimated as 60–90% from population-based and twin studies3,4. Both common and rare genetic variants are associated with the etiology of both disorders5,6,7,8. Neurodevelopmental mechanisms and related molecules are strongly involved in both SCZ and ASD9.
N-methyl-d-aspartate receptors (NMDARs) are ligand-gated ionotropic receptors that play important roles in synaptic plasticity within the central nervous system (CNS). NMDARs consist of two NR1 subunits in addition to either two NR2 (A, B, C, or D) and/or NR3 (A or B) subunits. Two of the four subunits must be NR1, and inclusion of the other two subunits results in multiple combinations10. Thus, combinations of subunits result in diversity of physiological and pharmacological functions. Long-term potentiation in the hippocampus depends on NMDAR activity11. NMDAR function is involved in CNS activity including cognitive function, behavior, and memory12,13. These functions are often impaired in psychiatric diseases such as SCZ and ASD. Therefore, NMDARs may contribute to the development of neurodevelopmental disorders.
A number of studies have shown that NMDAR antagonists (e.g., phencyclidine, ketamine) cause psychotic symptoms that are diagnostically difficult to differentiate from SCZ14. Moreover, NMDAR encephalitis, which was identified in 2007, has similar symptoms as SCZ. Autoantibodies against the NR1 subunit are produced in these patients, who develop diverse symptoms such as psychosis, memory deficits, seizures, and language disintegration15. Several studies suggest that NMDAR abnormalities exist in the CNS of SCZ patients16,17.
Many studies have reported that dysregulation of NMDARs is implicated in the etiology of SCZ. Mice with reduced expression of NR1 subunits display abnormal SCZ-like behavior that is ameliorated by treatment with antipsychotic drugs18. Interactions between the neuregulin 1 receptor (erbB4) and post-synaptic density (PSD)-95 are increased in these mice, and this interaction facilitates activation of erbB4, which suppresses NMDARs19. Moreover, d-serine, which acts as an agonist at NMDARs, has therapeutic effects when administered with certain antipsychotic drugs20. These findings consistently support the hypofunction hypothesis of NMDARs in SCZ.
Recent studies also suggest that NMDA is implicated in ASD21. In an animal model, both excessive and reduced NMDA dysfunctions result in ASD-like phenotypes. Shank2 is a scaffolding protein in the PSD and interconnects receptors at the PSD including NMDARs. Shank2−/− mice have reduced NMDAR activity and also exhibit ASD-like behaviors that are corrected by normalizing NMDAR functions22. IRSp53, which is also known as BAIAP2, acts as a scaffolding protein and regulates dendrite spines of excitatory synapses. In contrast to Shank2−/− mice, IRSp53−/− mice have enhanced NMDAR activity in the hippocampus, but also show impaired social interaction. These ASD-like behaviors are rescued by memantine. Thus, NMDAR dysregulation in the CNS may contribute to development of ASD23.
A recent genetic study that utilized next generation sequencing technology suggests that NMDARs are strongly implicated in the etiology of both SCZ and ASD. For instance, a large-scale genome-wide association study identified GRIN2A, which code NR2A, as an important candidate gene in SCZ susceptibility24. Other exome studies reported that de novo nonsense mutations and causative variants are found in GRIN2B, which code NR2B, in ASD25,26. In this context, we conducted a study to discover rare single nucleotide variants in GRIN2B, since several disease causative variants itself are identified in the same gene. Therefore, we sequenced the exonic regions of GRIN2B in SCZ and ASD to detect rare non-synonymous variants, and performed association studies targeting discovered variants in in the current study using large sample set of SCZ, ASD, and healthy controls.
Materials and Methods
Subjects
Two independent sample sets were used in this study. The first sample set, comprising 574 SCZ samples and 152 ASD samples, was used for rare missense or nonsense mutation screening. The second sample set, comprising 4145 SCZ patients, 381 ASD patients, and 4432 healthy controls (CON), was used for a genetic association study. Profiles (age and sex) of participants are shown in Table 1.
All participants were recruited in Nagoya University Hospital and its associated institutes and hospitals. All cases were diagnosed according to Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition. In this study, we enrolled patients with either SCZ or ASD. CON participants were evaluated with an unstructured interview to exclude individuals with a history of mental disorders. All participants were ethnically Japanese. Written informed consent was obtained from all subjects. All patients consented to participate in the study. When necessary, a patient’s ability to provide consent was confirmed by his or her guardian. Patients with communication difficulties, or those who did not consent were subsequently excluded. Ethical approval was granted by The Ethics Committee of the Nagoya University Graduate School of Medicine. All experiments were performed in accordance with the Committee’s guidelines and regulations.
Gene Screening and Variation Analyses
For each sample, we sequenced the coding region of GRIN2B (Chr12: 13,537,337–13,980,363 reverse strand, GRCh38 assembly, Ensembl transcriptID ENST00000609686). Genomic DNA was extracted from each subject’s peripheral blood or saliva with standard methods. Sequencing was performed with the standard capillary sequencing method. Primers for target amplification and the dye-termination reaction were designed using FastPCR (PrimerDigital Ltd. Helsinki, Finland) and PerlPrimer respectively27. Primer sequences are listed in Supplementary Table 1. Amplification primer was tested using UCSC in silico PCR (http://genome.ucsc.edu/cgi-bin/hgPcr) to verify that the intended targets were correct. We used the TAKARA LA Taq polymerase (Takara Bio Inc., Shiga, Japan) for PCR. After PCR amplification, amplicons were processed with Illustra Exonuclease I and Alkaline Phosphatase (GE Healthcare and Life Science, Little Chalfont, United Kingdom) to digest excess primers. The cycle sequencing reaction was then performed with the Big Dye Terminator v3.1 Cycle sequencing kit (Applied Biosystems, Foster City, CA, USA). The reaction products were purified with magnetic beads (CleanSEQ Kit, Beckman Coulter, Krefeld, Germany). Purified products were sequenced on a 3130xL Genetic Analyzer (Applied Biosystems).
Variant Reporter Software (Applied Biosystems) was used for mutation detection analysis. After all variants were screened, we narrowed it down the variants to perform follow-up analyses. Selection inclusion criteria was as follows. (1) Non-synonymous variants including nonsense single nucleotide variants (SNVs), missense SNVs, frame shifts, and small INDELs as candidate pathological variants. (2) We included only rare mutations (minor allele frequency <1%). In each case, the detected variations were validated in a discrete experiment at least once.
Bioinformatics Analyses
After variant selection, we analyzed variants using the following tools: (1) Human Protein Reference Database (http://www.hprd.org/index_html) and the Pfam database (http://pfam.xfam.org/)28 for confirming protein structure and localizations of variants. (2) Polyphen229, Sorting Tolerant from Intolerant (SIFT)30, and PMUT31 were used to evaluate functional changes arising as a consequence of alteration in amino acid sequence.
Genetic Association Analyses
For each adopted variant, case-control genotyping studies were performed. We used the Custom Taqman® SNP Genotyping assay (Applied Biosystems) for each variant. DNA samples were prepared on 384-microtiter plates with a positive control. PCR was performed with Taqman® Universal Master Mix 2 with UNG (Applied Biosystems). Allelic discrimination analyses were performed on an ABI PRISM HT7900 sequence detection system (Applied Biosystems). To determine if each detected sample did carry the variant of interest, positive samples were substantiated by at least one further experiment. Failed samples were also reassessed using a second experiment. We omitted samples in which we failed to detect the signals in two experiments.
For the association analysis, statistical tests were performed using JMP Pro (SAS Institute, Cary, NC, USA). Differences in allele and genotype frequencies of the mutations were compared between SCZ patients/controls, ASD patients/controls, and SCZ + ASD patients/controls respectively and significant was calculated using Fisher’s exact test (two-tailed). The threshold of significance set at p < 0.05.
Results
Mutation Screening
We identified five rare missense mutations and 14 synonymous mutations within GRIN2B coding exons (Table 2 and Supplementary Table 2).
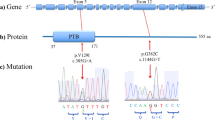
The first mutation was found in the N-terminal domain, the second was found in the ligand-gated ion channel domain, and the other three mutations were within the C-terminal (intracellular) domain (Fig. 1).

Location of missense variants.
(a) Variants with respect to the secondary structure of GRIN2B. (b) This image was created by referring to the crystal structure in the Protein Data Bank (http://www.rcsb.org/pdb/home/home.do. PDB ID: 4TLL)1). The location of each discovered variant (red star) is shown on the image. Columns colored deep blue, green, bright blue, and orange correspond to transmembrane helix1, p-loop, transmembrane helix2, and transmembrane helix3, respectively. Abbreviations: NTD, N-terminal domain; LBD, Ligand-binding domain; CTD, C-terminal domain.
We searched three genetic databases (dbSNP build143 (http://www.ncbi.nlm.nih.gov/SNP/index.html), 1000 Genome Project (http://www.1000genomes.org/), and Exome Variant Server (http://evs.gs.washington.edu/EVS/)) to determine if these missense mutations were novel variants. Three missense mutations (V18I, A590T, and G1040S) had already been registered on dbSNP. Additionally, genotype data for A590T were found on the Exome Variant Server. The aforementioned databases, as well as ExAC Browser (http://exac.broadinstitute.org), did not report R1099H and K1292R respectively.
Bioinformatics Analyses
The five missense variants were analyzed with Polyphen2, SIFT, and PMUT. The overall results are shown in Table 2 along with the screening result. According to the results of these analyses, R1099H was considered to have the most damaging effect due to the substitution in the protein; V18I and K1292R were thought to have a modest effect.
Results of Genetic Association Analyses
The result of the genotyping experiment is shown in Table 3. All variants except K1292R were detected in the CON group. K1292R was only detected in the SCZ group, and three additional SCZ samples were found to carry K1292R. Using Fisher’s exact test, none of the variants had a significant association with SCZ or ASD.
Discussion
In summary, we first sequenced the coding region of GRIN2B in 574 SCZ and 152 ASD samples using the Sanger sequencing method. Subsequently, we conducted association analysis with a secondary sample set. We detected five rare missense variants through mutation screening. We also sequenced parents’ sample of ASD in screening sample sets, however, we did not detect any de novo mutation. Moreover, we could not find significant associations between SCZ/ASD and variants investigated in this study. Although K1292R (Supplementary Result 1) was observed only in SCZ samples, evidence of statistical association, if any, must be obtained in larger population studies.
The amino acid sequence includes an important residue for binding of NR2B to Ca2+/calmodulin-dependent protein kinase II (CaMKII)32,33 (Supplementary Fig. 1). This interaction is key for autoregulation of CaMKII function and synaptic plasticity31. Stefan et al. previously examined the alteration of the association between NR2B and CaMKII by replacing NR2B residues. They reported that substitution of K (Lysine) 1292 with Q (Glutamine) reduces the affinity of NR2B for CaMKII. In our genetic study, we detected R instead of Q, but we could not examine the effect caused by R substitution. This NR2B-CaMKII protein interaction induces hippocampal long-term potentiation and spatial learning34. Of note, in the case of singleton SNVs (variants detected only in one individual or one family), performing association analysis with an extremely large sample size will be needed to achieve statistical significance but will be very difficult. However, the sole fact that the SNV was detected in only case samples is suggestive of its genetic relevance.
Limitations
There were several limitations in this study that may have influenced our results. Firstly, we used 574 SCZ and 152 ASD cases for mutation detection. Whilst we detected five rare missense mutations that may confer a genetic risk to SCZ/ASD development, due to the size of our screening samples, we may have limited our potential to identify other disease-causing variants. Specifically, the number of variants detected within GRIN2B in healthy subjects10, indicated that GRIN2B is among top 1% of the human genes that are least likely to carry functional genetic variation relative to the genome wide expectation given the amount of apparently neutral variation the gene has. Therefore, future studies may benefit from using much larger sample sizes to increase the likelihood of detecting a larger number of functionally relevant rare single nucleotide variants within GRIN2B.
Secondly, we applied stringent filtering criteria to control for multiple testing, because larger number of statistical tests (i.e. larger number candidate SNVs) equals lower (i.e. stringent) p-values which in turn translates into larger sample sizes required to obtain statistical evidence. Therefore, we chose to limit the number of candidate SNVs in order to have realistic chance for detecting statistical association using sample which we had available for this study. It is of note, however, that we have detected K1292R only in cases. Although, for this variant we could not obtain statistically significant results per se (i.e. P-value < 0.05), it is still relevant from biological point of view that this variant was not detected in controls. Considering that we had detected only few number of candidate variants in GRIN2B may be indication of its functional significance as the strong negative selection pressure upon amino acid sequence of GRIN2B may be related to relative scarcity of variants observed in general population.
Finally, after using all of our genotyping samples, we conducted a power calculation of the statistical power for K1292R35. The parameter settings were: High-risk allele frequency, 0.0004; Prevalence, 0.01; Genotype relative risk Aa, 3; Genotype relative risk AA, 9; D-prime, 1; Marker allele frequency, 0.0004; Number of cases: 4145; Control-case ratio, 1.0693; User-defined type I error rate, 0.05; User-defined power, 0.80. According to our power calculation based on the minor allele frequency of genotyping, approximately 9109 SCZ samples are needed to obtain 80% power. In other words, based on our study design, we can achieve 80% power if GRR is larger than 5 under multiplicative model, and we can achieve nearly 99% power if GRR is larger than 7 under same genetic model.
Conclusion
In conclusion, we found five rare missense mutations with our mutation screening. We also conducted genotyping for each detected variant. Whilst we did not find any significant association between any variant with SCZ or ASD, we found that K1292R was only present within the SCZ samples. Whilst K1292R was considered to confer a vulnerable effect for carriers, further genotyping, using a larger sample size, is needed to better understand its association with SCZ development. Moreover, to comprehensively assess impact of rare variants affecting NMDA signaling in case of psychiatric disorders, genes beyond GRIN2B should be evaluated in the future studies.
Additional Information
How to cite this article: Takasaki, Y. et al. Mutation screening of GRIN2B in schizophrenia and autism spectrum disorder in a Japanese population. Sci. Rep. 6, 33311; doi: 10.1038/srep33311 (2016).
References
Saha, S., Chant, D. & McGrath, J. A systematic review of mortality in schizophrenia: is the differential mortality gap worsening over time? Archives of general psychiatry 64, 1123–1131, 10.1001/archpsyc.64.10.1123 (2007).
Baird, G. et al. Prevalence of disorders of the autism spectrum in a population cohort of children in South Thames: the Special Needs and Autism Project (SNAP). Lancet 368, 210–215, 10.1016/S0140-6736(06)69041-7 (2006).
Cannon, T. D., Kaprio, J., Lonnqvist, J., Huttunen, M. & Koskenvuo, M. The genetic epidemiology of schizophrenia in a Finnish twin cohort - A population-based modeling study. Archives of general psychiatry 55, 67–74, 10.1001/archpsyc.55.1.67 (1998).
Lichtenstein, P. et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet 373, 234–239 (2009).
Gauglerl, T. et al. Most genetic risk for autism resides with common variation. Nat Genet 46, 881–885, 10.1038/ng.3039 (2014).
Buxbaum, J. D. Multiple rare variants in the etiology of autism spectrum disorders. Dialogues in clinical neuroscience 11, 35–43 (2009).
Girard, S. L. et al. Mutation burden of rare variants in schizophrenia candidate genes. PloS One 10, e0128988, 10.1371/journal.pone.0128988 (2015).
Stefansson, H. et al. Common variants conferring risk of schizophrenia. Nature 460, 744–U799, 10.1038/nature08186 (2009).
Lewis, D. A. & Levitt, P. Schizophrenia as a disorder of neurodevelopment. Annual review of neuroscience 25, 409–432, 10.1146/annurev.neuro.25.112701.142754 (2002).
Paoletti, P., Bellone, C. & Zhou, Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nature reviews. Neuroscience 14, 383–400, 10.1038/nrn3504 (2013).
Malenka, R. C. & Bear, M. F. LTP and LTD: an embarrassment of riches. Neuron 44, 5–21, 10.1016/j.neuron.2004.09.012 (2004).
Newcomer, J. W. & Krystal, J. H. NMDA receptor regulation of memory and behavior in humans. Hippocampus 11, 529–542, 10.1002/hipo.1069 (2001).
Malhotra, A. K. et al. NMDA receptor function and human cognition: the effects of ketamine in healthy volunteers. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology 14, 301–307, 10.1016/0893-133X(95)00137-3 (1996).
Jentsch, J. D. & Roth, R. H. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology 20, 201–225, 10.1016/S0893-133X(98)00060-8 (1999).
Dalmau, J., Lancaster, E., Martinez-Hernandez, E., Rosenfeld, M. R. & Balice-Gordon, R. Clinical experience and laboratory investigations in patients with anti-NMDAR encephalitis. The Lancet. Neurology 10, 63–74, 10.1016/S1474-4422(10)70253-2 (2011).
Watis, L., Chen, S. H., Chua, H. C., Chong, S. A. & Sim, K. Glutamatergic abnormalities of the thalamus in schizophrenia: a systematic review. Journal of neural transmission 115, 493–511, 10.1007/s00702-007-0859-5 (2008).
Pilowsky, L. S. et al. First in vivo evidence of an NMDA receptor deficit in medication-free schizophrenic patients. Mol Psychiatry 11, 118–119, 10.1038/sj.mp.4001751 (2006).
Mohn, A. R., Gainetdinov, R. R., Caron, M. G. & Koller, B. H. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell 98, 427–436 (1999).
Hahn, C. G. et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nature medicine 12, 824–828, 10.1038/nm1418 (2006).
Heresco-Levy, U. et al. D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol Psychiat 57, 577–585, 10.1016/j.biopsych.2004.12.037 (2005).
Uzunova, G., Hollander, E. & Shepherd, J. The role of ionotropic glutamate receptors in childhood neurodevelopmental disorders: autism spectrum disorders and fragile x syndrome. Current neuropharmacology 12, 71–98, 10.2174/1570159X113116660046 (2014).
Won, H. et al. Autistic-like social behaviour in Shank2-mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265, 10.1038/nature11208 (2012).
Lee, E. J., Choi, S. Y. & Kim, E. NMDA receptor dysfunction in autism spectrum disorders. Current opinion in pharmacology 20, 8–13, 10.1016/j.coph.2014.10.007 (2015).
Schizophrenia Working Group of the Psychiatric Genomics, C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427, 10.1038/nature13595 (2014).
O’Roak, B. J. et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet 43, 585–589, 10.1038/ng.835 (2011).
De Rubeis, S. et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215, 10.1038/nature13772 (2014).
Marshall, O. J. PerlPrimer: cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics 20, 2471–2472, 10.1093/bioinformatics/bth254bth254 [pii] (2004).
Finn, R. D. et al. Pfam: the protein families database. Nucleic acids research 42, D222–D230, 10.1093/nar/gkt1223 (2014).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nature methods 7, 248–249, 10.1038/nmeth0410-248 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols 4, 1073–1081, 10.1038/nprot.2009.86 (2009).
Ferrer-Costa, C. et al. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics 21, 3176–3178, 10.1093/bioinformatics/bti486 (2005).
Bayer, K. U., De Koninck, P., Leonard, A. S., Hell, J. W. & Schulman, H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411, 801–805, 10.1038/35081080 (2001).
Bayer, K. U. et al. Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. The Journal of neuroscience: the official journal of the Society for Neuroscience 26, 1164–1174, 10.1523/JNEUROSCI.3116-05.2006 (2006).
Zhou, Y. et al. Interactions between the NR2B receptor and CaMKII modulate synaptic plasticity and spatial learning. The Journal of neuroscience: the official journal of the Society for Neuroscience 27, 13843–13853, 10.1523/JNEUROSCI.4486-07.2007 (2007).
Purcell, S., Cherny, S. S. & Sham, P. C. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 19, 149–150, DOI 10.1093/bioinformatics/19.1.149 (2003).
Acknowledgements
This work was supported by research grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan; the Ministry of Health, Labor and Welfare of Japan; a Grant-in-Aid for “Integrated research on neuropsychiatric disorders” carried out under the Strategic Research Program for Brain Sciences by the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Author information
Authors and Affiliations
Contributions
Y.T., T.K., C.W. and B.A. Conception and design of study, sample and data collection, data analysis and interpretation, and manuscript writing; H.K., J.X., I.K., K.I., D.M., M.S., A.Y., Y.A., T.O.-I., Y.N., S.K., Y.U., T.O. and N.O. Sample collection and data interpretation; M.I., M.A., N.T., Y.I., M.Y., H.N., H.U., J.E., H.K., T.S., T.Y. and N.I. Sample and data collection.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Takasaki, Y., Koide, T., Wang, C. et al. Mutation screening of GRIN2B in schizophrenia and autism spectrum disorder in a Japanese population. Sci Rep 6, 33311 (2016). https://doi.org/10.1038/srep33311
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep33311
This article is cited by
-
Single-locus and Haplotype Associations of GRIN2B Gene with Autism Spectrum Disorders and the Demographic and Clinical Characteristics of Patients in Guilan, Iran
Journal of Autism and Developmental Disorders (2024)
-
Targeting NMDA receptors in neuropsychiatric disorders by drug screening on human neurons derived from pluripotent stem cells
Translational Psychiatry (2022)
-
The GluN2B-Trp373 NMDA Receptor Variant is Associated with Autism-, Epilepsy-Related Phenotypes and Reduces NMDA Receptor Currents in Rats
Neurochemical Research (2022)
-
Rare loss of function mutations in N-methyl-d-aspartate glutamate receptors and their contributions to schizophrenia susceptibility
Translational Psychiatry (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
