
Abstract
The compound BaZn2Si2O7 shows a high coefficient of thermal expansion up to a temperature of 280 °C, then a transition to a high temperature phase is observed. This high temperature phase exhibits negative thermal expansion. If Ba2+ is successively replaced by Sr2+, a new phase with a structure, similar to that of the high temperature phase of BaZn2Si2O7, forms. At the composition Ba0.8Sr0.2Zn2Si2O7, this new phase is completely stabilized. The crystal structure was determined with single crystal X-ray diffraction using the composition Ba0.6Sr0.4Zn2Si2O7, which crystallizes in the orthorhombic space group Cmcm. The negative thermal expansion is a result of motions and distortions inside the crystal lattice, especially inside the chains of ZnO4 tetrahedra. Dilatometry and high temperature X-ray powder diffraction were used to verify the negative thermal expansion. Coefficients of thermal expansion partially smaller than −10·10−6 K−1 were measured.
Similar content being viewed by others

Introduction
Most of all materials expand during heating1,2,3. There are just a few silicates, exhibiting negative thermal expansion, such as beta-quartz and beta-eucryptite4,5,6. Besides, ZrW2O8 is one of the most commonly known negative thermal expansion (NTE) materials exhibiting a coefficient of thermal expansion of −8.7·10−6 K−1 7,8. By contrast, barium silicates, normally exhibit extremely high coefficients of thermal expansion9. The compound BaZn2Si2O7 shows a high coefficient of thermal expansion up to a temperature of 280 °C, then a transition to a high temperature phase is observed, running parallel to a steep increase in volume of around 2%10. Below the phase transition, where the phase has a monoclinic structure with the space group C2/c, the mean value of the coefficient of thermal expansion is around 17.6·10−6 K−1 11,12. Above this phase transition, the phase has the orthorhombic space group Ccm21 and very low and partially negative thermal expansion is observed11,12. The stabilization of this high temperature phase down to room temperature was not possible until now. We found a new family of materials with giant negative coefficients of thermal expansion, partially smaller than −10·10−6 K−1, in solid solutions of the system Ba1−xSrxZn2Si2O7. If Ba2+ is successively replaced by Sr2+, a new phase with a structure, similar to that of the high temperature phase of BaZn2Si2O7, forms. At the composition Ba0.8Sr0.2Zn2Si2O7, this new phase is completely stabilized and can easily be crystallized from silicate glasses which is of great importance because all commercial available zero expansion materials are glass-ceramics. To the best of our knowledge, it is the first new silicate phase with negative thermal expansion reported in the past 50 years. In comparison to alumosilicates, the manufacturing process should be much easier.
Ceramics and glass-ceramics containing BaZn2Si2O7 and other isostructural compounds are well known as high thermal expansion materials10,11,12,13. The substitution of Zn2+ by other ions with similar ionic radii and the same valence state, such as Co2+, Mg2+ or Ni2+, leads to a shift of the phase transition towards higher temperatures. Klasens et al. reported already in 195715 that the substitution of BaO by SrO leads to the formation of a new phase with unknown crystal structure. They also reported that a complete replacement of BaO by SrO is not possible. Up to now, this phase was not further studied, especially it was not discovered that this phase has a negative thermal expansion.
In Fig. 1, the crystal structure of the Ba0.6Sr0.4Zn2Si2O7 phase, determined using single crystal X-ray diffraction is shown in the left part of the image in four different views. The samples were prepared from a stoichiometric melt with a BaO:SrO-ratio of 1:1, but the refinement of the crystal structure showed that the composition of the investigated single crystal is Ba0.6Sr0.4Zn2Si2O7. This composition was also confirmed via energy-dispersive X-ray spectroscopy (EDX). The reason for this difference should be a not completely homogenized melt from which these single crystals were prepared. A preferred incorporation of Ba2+ into the structure is not assumed because of the much higher ionic radius in comparison to Sr2+.

The crystal structure of the Ba0.6Sr0.4Zn2Si2O7 phase at 20 °C.
The left part of the image displays the crystal structure with different views. On the right side, a comparison between the crystal structures of Ba0.6Sr0.4Zn2Si2O7 and HT-BaZn2Si2O7 is displayed. The blue spheres are the Ba2+ and Sr2+ ions inside the Ba0.6Sr0.4Zn2Si2O7 phase. In the case of BaZn2Si2O7, the blue balls only describe the positions of Ba2+.
The crystal structure information as well as the refinement parameters are summarized in Table 1. Silicon as well as zinc are fourfold coordinated. Two SiO4 tetrahedra are bridged by an oxygen. In analogy, also the ZnO4 tetrahedra are bridged by oxygen. In these tetrahedra, two of four oxygens act as bridges between two ZnO4 tetrahedra and one SiO4 tetrahedron. The other two oxygens bridge only two silicon atoms. The Ba- and Sr- atoms are surrounded by 5 oxygens, forming a pyramid with a quadratic basal plane. The right part of Fig. 1 shows a comparison of the b-c-plane of Ba0.6Sr0.4Zn2Si2O7 and the a-c-plane of HT-BaZn2Si2O7. The image of the latter was calculated from the crystal structure information of HT-BaZn2Si2O7 measured at 350 °C in ref. 14. The different definitions of the unit cells and lattice parameters of both phases make it necessary to compare different planes and directions as it is done in Fig. 1 for the b-c- and the a-c-planes. It can be seen that there are similar atomic arrangements in both phases (lower part on the right side of Fig. 1). The differences in the crystal structures of both phases are only marginal so that both phases should exhibit similar thermal expansion properties.
In Fig. 2, X-ray powder diffraction patterns in the solid solution series Ba1-xSrxZn2Si2O7 with different values of x, measured at room temperature, are shown. It can be seen that the crystal structure changes from x = 0 to x = 0.1. The diffractogram with x = 0 is that of pure LT- BaZn2Si2O7. In the diffraction pattern with x = 0.1, it can be seen that the new crystal phase has formed. Minor concentrations of crystals with the structure of LT-BaZn2Si2O7 were also found. At higher x-values, i.e. higher Sr-concentrations, the new structure is completely stabilized and hence, the only occurring. The peak positions for 0.1 ≤ × ≤ 0.9 are approximately in agreement with those from the single crystal measurement and those from ref.15 (see the lower two patterns on the left side of Fig. 2). The peak positions of the respective graphs are somewhat shifted; the introduction of more strontium into the lattice results in somewhat larger 2θ values, i.e. in smaller lattice constants, which can be clearly seen by comparing the samples with x = 0.1 and 0.9. This can easily be explained by the smaller cation radius of Sr2+. A complete substitution of BaO by SrO does not result in single phase materials but in a mixture of crystals with the structure of HT-BaZn2Si2O7, together with Sr2ZnSi2O7 and Zn2SiO4. However, in the range of 0.1 ≤ × ≤ 0.9 single phase materials with the crystal structure similar to HT-BaZn2Si2O7 were obtained.

Changes in the crystal structure and the thermal expansion behavior in the solid solution series Ba1-xSrxZn2Si2O7 for different values of x.
On the left side, X-ray powder diffraction patterns measured at room temperature are presented. In the lower part of the graph, the peak positions, determined by Klasens et al. in 195711 (A), those calculated from the crystal structure of a Ba0.6Sr0.4Zn2Si2O7 single crystal (B) and those of LT-BaZn2Si2O7 taken from ref. 13 (C) are given. In the upper part, measured diffractograms with x = 0, 0.1 and 0.9 are shown. On the right side, the thermal expansion behavior, determined via dilatometry, is given for x = 0, 0.1 and 0.8. The curve of the sample with x = 0 was taken from Ref. 10.
The change in the crystal structure runs parallel to an exceptional change in the thermal expansion behavior. This can be seen in the right part of Fig. 2. The sample with x = 0 exhibits extremely high thermal expansion behavior (the data were taken from ref. 10). At around 280 °C, a phase transition leads to the formation of the HT-modification, which shows partly negative thermal expansion. The samples with x = 0.1 and 0.8, where a crystal structure similar to HT-BaZn2Si2O7 is stable, show extremely low thermal expansion behavior, which is comparable to that of the sample with x = 0 above around 300 °C. It should be mentioned that in the case of NTE materials, the thermal expansion behavior measured by dilatometry, strongly depends on the sample preparation and microstructure 16,17. The sintered ceramic bodies showed strong hysteresis behavior, which might be affected by micro cracking. However, all samples with the structure of HT-BaZn2Si2O7 exhibit negative thermal expansion. A correlation of the dilatometric thermal expansion and the SrO concentration was not found.
A contraction during heating can also be seen in Fig. 3, which pictorially presents the changes in the crystal structure with temperature. The upper part of the picture (A) shows a rotational movement of ZnO4 tetrahedra inside a ZnO4 chain. This rotation in general leads to a strong increase in length in the direction of the chain (see the length, denoted as “x” in Fig. 3A) as well as a small increase perpendicular to this direction (y). However, as it is illustrated by the dashed lines, the rotation of the tetrahedra also leads to a strong shortening of the distance z. In the direction perpendicular to the chain (b-axis), the ZnO4 tetrahedra are connected with rigid SiO4 tetrahedra (Fig. 3B). A rotation inside the chain now leads to the effect that some of the SiO4 tetrahedra are pulled near the chain and other are slightly pushed away. As it is illustrated in Fig. 3A, the effect of pulling, i. e. shortening in the direction of the b-axis, is much more pronounced than the contrary effect. This leads to strong distortions of the tetrahedra, which are much more pronounced for the ZnO4 tetrahedra, which will be strongly compressed, perpendicular to the chain and can be seen in Fig. 3C in the a-b-plane of the crystal. These results are based on single crystal X-ray diffraction measurements. On the left side, the crystal structure is displayed. On the right side, the distortions of the SiO4 and the ZnO4 tetrahedra are shown for different temperature ranges. It can be clearly seen that the distortion of the ZnO4 tetrahedra is much stronger than that of the SiO4 tetrahedra. This distortion, together with small tilting motions of the tetrahedra lead to an obvious decrease in the O1–O1 distance of 0.652% between −55 and 80 °C. This is the most pronounced contraction found in this crystal structure. Furthermore, the rotation of ZnO4 units also leads to a change in bond angles away from that of an ideal tetrahedron. As it is demonstrated in Fig. 3B, the SiO4 tetrahedra will be stretched in the direction of the chain, which can be seen by the increase of the respective O-Si-O angle, which increases from 115.778 at −55 °C to 115.921 at 80 °C.

The origin of the negative thermal expansion in crystals with the structure of Ba0.6Sr0.4Zn2Si2O7.
(A) Movement of ZnO4-tetrahedra in a single chain. (B) Rotation and distortion inside the network. (C) Distortions of SiO4- and ZnO4-tetrahedra at different temperatures. The values were taken from single crystal X-ray diffraction measurements. On the left side, the a-b-plane of one unit cell is shown. The red arrows between two O1-atoms show the main direction of shrinkage with a shortening of 0.652% during heating from −55 °C to 80 °C. On the right side, the changes in the O-Si bond length (I, II) and the changes in the O-Si-O bond angles (III, IV) are displayed for two different temperature ranges, given in [%] and [°], respectively. By analogy it was done for the [ZnO4]2−-tetrahedra (V-VIII).
Furthermore, the rotational movement of ZnO4 tetrahedra will also explain the highly anisotropic thermal expansion behavior with coefficients of thermal expansion of 9.5∙10−6 K−1 (a-axis), −32.1∙10−6 K−1 (b-axis) and 23.1∙10−6 K−1 (c-axis) (measured between 30 and 600 °C). The highest thermal expansion can be found in the direction of the ZnO4 chains and negative thermal expansion perpendicular to the chain in the direction of the b-axis. The thermal expansion in the direction of the a-axis does not seem to be affected by the motions of the chains. This reason can be found in the left part of Fig. 1, where the a-c-plane is shown. There it can be seen that the ZnO4 chains are highly oriented in this plane so that a motion of the chains in these directions should be energetically not favored.
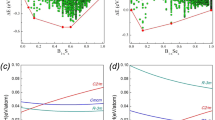
Figure 4 shows the thermal expansion behavior of Ba0.6Sr0.4Zn2Si2O7. Powdered specimens of Ba0.6Sr0.4Zn2Si2O7 were studied using high temperature powder X-ray diffraction. For a better illustration, Fig. 4 shows the powder XRD-patterns for a narrow 2θ-range from 18 to 42° (left part of the picture). To enable a more accurate determination of the 2θ-values, the powder was mixed with Al2O3 as internal standard. Furthermore, the peak positions of the Ba0.6Sr0.4Zn2Si2O7 phase and Al2O3, taken from the single crystal measurement and Ref. 18are shown. It is seen that the peak at around 27° is shifted to larger 2θ values with increasing temperature, which shows that the corresponding lattice spacings decrease with increasing temperature. This peak can be attributed to the (220) lattice plane. In the upper right corner of Fig. 4, the cell parameters, calculated from these XRD measurements, are shown as a function of temperature. A strong contraction of the lattice parameter of the b-axes with increasing temperature is obvious. This effect leads to the contraction of the complete unit cell.

Thermal expansion of Ba0.6Sr0.4Zn2Si2O7.
On the left side, XRD-patterns, recorded at various temperatures are presented in a 2θ range between 18° and 42°. In the lower two diffractograms, the peak positions of Al2O3 (internal standard) and Ba0.6Sr0.4Zn2Si2O7 are shown. The peaks of Al2O3 were taken from ref. 14. In the upper right corner, the cell parameters, determined by X-ray powder diffraction are given as a function of the temperature. In the lower right corner, the dilatometric thermal expansion behavior of bulk ceramics, prepared with different sintering conditions is illustrated: (A) sintered at 1300 °C without compression (B) isostatically pressed with 100 bar and afterwards sintered at 1175 °C; (C) hot pressed at 1100 °C with an uniaxial pressure of 28 MPa; (D) sintered at 1270 °C without compression. Furthermore, the volume of the unit cell is shown (blue symbols).
However, as mentioned above, the coefficient of thermal expansion of the pure crystalline ceramics strongly depends on the sample preparation. The strong anisotropic thermal expansion behavior may lead to the formation of cracks, additionally lowering the coefficient of thermal expansion. Hence, the thermal expansion behavior determined via HT-XRD measurements, is the more reliable and might differ strongly from dilatometric results. An illustration of the NTE behavior of samples with the composition Ba0.5Sr0.5Zn2Si2O7 prepared at different temperatures and sintering conditions is shown in the lower right corner of Fig. 4, where the coefficient of thermal expansion in a temperature range between 100 and 800 °C varies between −4.3 and −14.8∙10−6 K−1. The strong variation of the thermal expansion behavior of bulk ceramic samples in dependence of the preparation method is commonly known and the fabrication of dense sintered and crack-free NTE materials is highly challenging19,20. This problem is avoided by crystallization of such phases from glasses.
Crystalline phases with negative thermal expansion coefficients are not only suitable for the production of materials with a coefficient of thermal expansion, which is negative or close to zero, but can also be used for a large variety of other purposes. Among these are:
-
Materials composed by a bulk material with positive thermal expansion and a surface layer of a material with negative thermal expansion. During cooling of such a material, compressive stresses at the surface are formed which might result in high strength materials.
-
Lamellar structures composed of alternating layers of materials with positive and negative thermal expansion. Also here, stresses are formed during cooling which should especially result in high fracture toughness.
-
Joining of materials with low thermal expansion, such as vitreous silica or Ceran© Glass Ceramics. The seal has an adapted thermal expansion coefficient and can be sealed at temperatures notably below the softening point of e.g. Ceran© or vitreous silica. Such seals up to now are not available.
-
Sealing Materials with adjustable thermal expansion properties, in order to seal a large variety of materials, such as metals with comparatively high coefficients of thermal expansion used in high-temperature reactors or materials with lower coefficients of thermal expansion such as glasses or ceramics.
Methods
Ternary silicates from the solid solution series Ba1-xSrxZnSi2O7 (0 ≤ × < 1) were synthesized via conventional ceramic mixed oxide route so that 25 g of ceramic powder were obtained. Mixtures of SiO2 (Carl Roth GmbH + Co. KG, >99%), BaCO3 (VK Labor- und Feinchemikalien, pure), SrCO3 (Ferak Berlin, >99%) and ZnO (Carl Roth, >99%) were used as raw materials. The mixtures were heated up to temperatures between 1125 to 1250 °C, kept for 30 to 50 h with several intermediate regrinding steps.
The phase purity was verified by X-ray powder diffraction (XRD), using a SIEMENS D5000 Bragg-Brentano diffractometer and CuKα radiation. The XRD-patterns of the powdered samples were recorded between 10 ≤ 2θ ≤ 60° with a step size of Δθ = 0.02° and a counting time of 1 s per step.
The same device was used for high temperature XRD (HT-XRD). Here, the diffractometer was equipped with an ANTON PAAR HTK 10 heating stage. The powdered samples were heated to the respective temperatures, kept for several minutes until temperature equilibrium is reached and afterwards an XRD scan was performed. The samples were measured in a 2θ range from 10–60°. All the samples, analyzed by HT-XRD contained 33 wt% of α-Al2O3 as an internal standard. The peak positions and cell parameters of α-Al2O3 are known from the ICSD database (27 °C: ICSD10425, 300 °C: ICSD160605, 600 °C: ICSD160606, 900 °C: ICSD160607) and agree with other data from the literature18,21. From these patterns, the cell parameters were calculated with the software TOPAS from Bruker.
Single crystals were prepared using the following procedure: Ba0.5Sr0.5Zn2Si2O7 ceramic powder was melted in a small platinum crucible (6 mm in diameter and 2 mm in height) at around 1500 °C and slowly cooled down to 1150 °C with a cooling rate of 1 K/min. Afterwards, the sample was cooled fast to room temperature. The so prepared sample was polycrystalline, but contained large single crystalline regions. Single crystals were obtained by mechanically destroying the polycrystals. A polarization microscope was used to identify the single crystals. After single crystal X-ray diffraction, the composition of the crystal was determined using energy dispersive X-ray spectroscopy (EDS). Therefore, a JEOL JSM 7001F electron microscope was used. The composition of the crystal was determined using an acceleration voltage of 30 kV. This high voltage has to be used in order to stimulate the Sr Kα energy at 14.163 keV. Lower values lead to erroneous values because of the strong overlapping of the Kα energy of Si at 1.74 keV and the Lα energy of Sr at 1.806 keV.
Dilatometric measurements were performed between room temperature and 900 °C (heating rate 5 K/min), using a dilatometer NETZSCH Dil 402 PC. The ceramic powders were cold isostatically pressed into cylindrical shape (diameter: 6–8 mm, length: 5–25 mm) and afterwards sintered between 1125 and 1300 °C. The hot pressed sample was prepared by uniaxial pressing with a pressure of 28 MPa, kept for 5 min at 1100 °C.
The single-crystal structure determination was performed as follows. The intensity data for the compounds were collected on a Nonius KappaCCD diffractometer using graphite-monochromated Mo-Kα radiation. The data were corrected for Lorentz and polarization effects; absorption was taken into account on a semi-empirical basis using multiple-scans22,23,24.
The structures were solved by direct methods (SHELXS25) and refined by full-matrix least squares techniques against Fo2 (SHELXL-9725). The Ba2+ and Sr2+ positions are disordered on the m2 m site symmetry of the space group Cmcm (No. 63). The occupancy factors were refined for the same crystal at different temperatures for Sr2+ to 39.8(7) % (220 K), 39.6(7) % (293K) and 40.5(7) % (353 K), respectively. For the final refinement, the site occupancy factors were fixed for all temperatures to 60% for Ba2+ and to 40% for Sr2+. All atoms were refined anisotropically25.
Crystallographic data as well as structure solution and refinement details are summarized in Table 1. DIAMOND 3.2 was used for structure representations.
Additional Information
How to cite this article: Thieme, C. et al. Ba1−xSrxZn2Si2O7 - A new family of materials with negative and very high thermal expansion. Sci. Rep. 5, 18040; doi: 10.1038/srep18040 (2015).
References
Zheng, X. G. et al. Giant negative thermal expansion in magnetic nanocrystals. Nature Nanotech. 3, 724–726 (2008).
Takenaka, K. Negative thermal expansion materials: technological key for control of thermal expansion. Sci. Technol. Adv. Mater. 13, 013001 (2012).
Grima, J. N., Zammit, V. & Gatt, R. Negative thermal expansion. Xjenza 11, 17–29 (2006).
Ernst, G., Broholm, C., Kowach, G. R. & Ramirez, A. P. Phonon denstity of states and negative thermal expansion in ZrW2O8 . Nature 396, 147–149 (1998).
Müller, G. & Sternitzke, M. Computer modelling of structure and thermal expansion of β-quartz-and keatite-type aluminosilicates. J. Mater. Sci. Lett. 12, 278–280 (1993).
Gillery, F. H. & Bush, E. A. Thermal Contraction of beta-Eucryptite (Li2O.Al2O3.2SiO2) by X-Ray and Dilatometer Methods. J. Am. Ceram. Soc. 42, 175–177 (1959).
Evans, J. S. O., Mary, T. A., Vogt, T., Subramanian, M. A. & Sleight, A. W. Negative thermal expansion in ZrW2O8 and HfW2O8 . Chem. Mater. 8, 2809–2823 (1996).
Lommens, P. et al. Synthesis and thermal expansion of ZrO2/ZrW2O8 composites. J. Eur. Ceram. Soc. 25, 3605–3610 (2005).
Kerstan, M. & Rüssel, C. Barium silicates as high thermal expansion seals for solid oxide fuel cells studied by high-temperature X-ray diffraction (HT-XRD). J. Power Sources 196, 7578–7584 (2011).
Kerstan, M., Thieme, C., Grosch, M., Müller, M. & Rüssel, C. BaZn2Si2O7 and the solid solution series BaZn2−xCoxSi2O7 (0≤x≤2) as high temperature seals for solid oxide fuel cells studied by high-temperature X-ray diffraction and dilatometry. J. Solid State Chem. 207, 55–60 (2013).
Kerstan, M., Müller, M. & Rüssel, C. Thermal expansion of Ba2ZnSi2O7, BaZnSiO4 and the solid solution series BaZn2−xMgxSi2O7 (0≤x≤2) studied by high-temperature X-ray diffraction and dilatometry. J. Solid State Chem. 188, 84–91 (2012).
Pascual, M. J. et al. Transport properties of sealants for high-temperature electrochemical applications: RO-BaO-SiO2 (R = Mg, Zn) glass-ceramics. J. Eur. Ceram. Soc. 26, 3315–3324 (2006).
Thieme, C. & Rüssel, C. J. Mater. Sci. 10.1007/s10853-015-9398-x (2015).
Lin, J. H. et al. Phase transition and crystal structures of BaZn2Si2O7 . J. Phys. Chem. Solids 60, 975–983 (1999).
Klasens, H. A., Hoekstra, A. H. & Cox, A. P. M. Ultraviolet Fluorescence of Some Ternary Silicates Activated with Lead. J. Electrochem. Soc. 104, 93–100 (1957).
Chu, C. N., Saka, N. & Suh, N. P. Negative thermal expansion ceramics: A review. Mater. Sci. Eng. 95, 303–308 (1987).
Pelletant, A. et al. Grain size dependence of pure β-eucryptite thermal expansion coefficient. Mater. Lett. 66, 68–71 (2012).
Ishizawa, N., Miyata, T., Minato, I., Marumo, F. & Iwai, S. A structural investigation of α-Al2O3 at 2170 K. Acta Cryst. B 36, 228–230 (1980).
Benavente, R. et al. Fabrication of near-zero thermal expansion of fully dense β-eucryptite ceramics by microwave sintering. Ceram. Int. 40, 935–941 (2014).
Roy, R., Agrawal, D. K. & McKinstry, H. A. Very Low Thermal Expansion Coefficient Materials. Annu. Rev. Mater. Sci. 19, 59–81 (1989).
Kondo, S., Tateishi, K. & Ishizawa, N. Structural Evolution of Corundum at High Temperatures. Jpn. J. Appl. Phys. 47, 616–619 (2008).
Hooft, R. W. W. (1998). COLLECT: Data Collection Software. Nonius BV, Delft, The Netherlands. URL http://www.nonius.nl.
Otwinowski, Z. & Minor, W. Processing of X-Ray Diffraction Data Collected in Oscillation Mode in Methods in Enzymology, Vol. 276 (eds. Carter, W. C. et al.) 307–326 (Academic Press, 1997).
Bruker (2002). SADABS 2.10. Bruker-AXS Inc., Madison, USA. URL http://www.bruker.com.
Sheldrick, G. M. Foundations of Crystallography. Acta Cryst. A64, 112–122 (2008).
Author information
Authors and Affiliations
Contributions
C.T. prepared all figures and performed all measurements excluding the single-crystal X-ray diffraction. H.G. was responsible for the single crystal X-ray measurements and prepared Table 1. He also prepared the .cif-file submitted to the ICSD database. C.R. was responsible for the coordination of the experiments. All authors participated in writing and reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Thieme, C., Görls, H. & Rüssel, C. Ba1−xSrxZn2Si2O7 - A new family of materials with negative and very high thermal expansion. Sci Rep 5, 18040 (2016). https://doi.org/10.1038/srep18040
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep18040
This article is cited by
-
Elastic mechanics solution of thermal expansion of bi-material curved beam and its application to negative thermal expansion metamaterials
Scientific Reports (2022)
-
Noble metals Pt, Au, and Ag as nucleating agents in BaO/SrO/ZnO/SiO2 glasses: formation of alloys and core–shell structures
Journal of Materials Science (2022)
-
Effect of isothermal treatment on the structural, microstructure, and physio-mechanical properties of Li2O-Al2O3-SiO2 glass–ceramic
Journal of the Australian Ceramic Society (2022)
-
Negative thermal expansion in porous glass-ceramics based on Mg2Al2B2Si5O18 prepared from Saudi raw materials
Journal of Mechanical Science and Technology (2020)
-
Crystal growth velocities of a highly anisotropic phase obtained via surface and volume crystallization of barium–strontium–zinc silicate glasses
Journal of Materials Science (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
