
Abstract
Long intergenic non-coding RNAs (lincRNAs) play important roles in many cellular processes. Here, we present the first systematic identification and characterization of lincRNAs in fetal porcine skeletal muscle. We obtained a total of 55.02 million 90-bp paired-end reads and assembled 54,550 transcripts using cufflinks. We developed a pipeline to identify 570 multi-exon lincRNAs by integrating a set of previous approaches. These putative porcine lincRNAs share many characteristics with mammalian lincRNAs, such as a relatively short length, small number of exons and low level of sequence conservation. We found that the porcine lincRNAs were preferentially located near genes mediating transcriptional regulation rather than those with developmental functions. We further experimentally analyzed the features of a conserved mouse lincRNA gene and found that isoforms 1 and 4 of this lincRNA were enriched in the cell nucleus and were associated with polycomb repressive complex 2 (PRC2). Our results provide a catalog of fetal porcine lincRNAs for further experimental investigation of the functions of these genes in the skeletal muscle developmental process.
Similar content being viewed by others

Introduction
In mammals, a large proportion of the genome is composed of intergenic regions, yet little was known about the transcription of these regions at the time of completion of the human genome1. Several studies in the past decade, however, have revealed that most of these regions may represent novel transcribed regions2,3,4, where transcripts longer than 200 nucleotides in length are localized. Much of this newly discovered major class of transcripts has very weak or no protein-coding potential and, thus, was defined as long intergenic non-coding RNAs (lincRNAs)5. Since the identification of the first two imprinted lincRNAs, H19 and Xist (X inactive specific transcript), in the early 1990s6,7, lincRNAs have emerged as an exciting new molecules with potential roles in a variety of cellular processes, including gene regulation8,9, X-chromosome inactivation7,10,11, reprogramming12, pluripotency maintenance13, embryonic development14 and paraspeckle formation15,16.
In recent decades, the main goal of pig breeding has been to improve the pig growth rate and muscularity17. Several studies have indicated that postnatal skeletal muscle growth is largely affected by prenatal skeletal muscle development18,19. Therefore, understanding the network dynamics of the muscle transcriptome during earlier fetal stages will be of great importance in unraveling the complex mechanism underlying muscle development. The vast majority of transcripts behave as non-coding RNAs (ncRNAs)20,21,22, which include a large portion of lincRNAs23,24. However, previous studies on porcine fetal skeletal muscle have primarily focused on protein-coding genes25,26,27,28 and miRNAs29,30,31,32 instead of lincRNAs; consequently, transcriptional information for skeletal muscle growth in swine is incomplete. Of note, previous investigations found that polycomb repressive complex 2 (PRC2) plays a key role in regulating myogenesis33 and that approximately 20% of lincRNAs are bound by PRC234. These findings indicate that lincRNAs are most likely involved in the development of skeletal muscle.
Thousands of lincRNAs have been identified in humans and mice5,35, some of which demonstrate strong evolutionary signals of inter-species conservation5. However, the assembly of porcine lincRNA sequences using conserved lincRNAs as seeds for the collection of expressed sequence tags (ESTs) is still laborious and ineffective36. In particular, lincRNA transcripts obtained from different sources make it difficult to study their roles in skeletal muscle development. More recently, RNA-seq technology37 and computational methods developed for transcriptome reconstruction35,38 have facilitated comprehensive gene annotation and functional characterization of lincRNAs. These approaches have been successfully applied to identify and characterize lincRNAs in a given tissue or cell line39,40,41.
Muscle development in pig fetuses involves two major waves of fiber generation: primary fiber formation at 35–60 days post coitus (dpc) and secondary fiber formation at 54–90 dpc42. However, some studies have shown that the development of muscle fibers in the fetal pig is mostly complete by 70–75 dpc43,44, which is consistent with additional recent findings that porcine myogenesis is almost complete before 77 dpc28. Moreover, it was shown that the stage ranging from 50 to 75 dpc is critical for the formation of various muscle phenotypes28. Thus, the 50–75 dpc period is a critical stage of fetal skeletal muscle development.
In this study, we report the systematic identification and characterization of lincRNAs in porcine fetal skeletal muscle from paired-end RNA-seq data that were obtained from a pool of samples at 50, 55, 60, 65 and 75 dpc. We further characterized the basic features of a conserved mouse lincRNA, including its subcellular localization and its association with chromatin-modifying complexes. Our study paves the way for further studies exploring the functional roles of lincRNA during porcine skeletal muscle development.
Results
Read mapping and transcript assembly
A total of 55.02 million 90-bp pair-end reads were obtained after filtering out low-quality reads and removing the adaptor sequences. Approximately 73.1% of the total clean reads were mapped to the Sus scrofa genome assembly 10.2 and 54,550 assembled transcripts were produced.
Genomic information of porcine lincRNAs
We developed a highly stringent filtering pipeline (Figure 1) to identify porcine lincRNAs using an integrated experimental and computational approach. In total, our pipeline yielded 570 lincRNA transcripts, corresponding to 476 lincRNA genes. Some lincRNA genes were alternatively spliced, containing 1.2 isoforms per lincRNA locus on average. We found that 45.4% of the total lincRNA was transcribed near (<10 kb) known protein-coding genes. The average size of porcine lincRNA was approximately 1,043 bp, with a range of 200 to 834 nucleotides that span 2.5 exons on average, which is similar to that of human lincRNA39. The average exon length of lincRNA was 417 bp and lincRNAs that contained two exons accounted for 65.7% of the total lincRNAs. We also found that our lincRNA genes contained canonical splice sites (GT-AG); these lincRNAs were distributed in all chromosomes except the Y chromosome.

Overview of the stringent filtering pipeline used to identify the resulting 570 lincRNAs.
At each step, the vertical arrow denotes the transcripts that passed the filter and the box denotes those that were removed.
Comparison between lincRNAs and protein-coding genes
We also obtained 14,836 protein-coding transcripts that corresponded to 12,372 genes (an average of 1.2 isoforms per protein gene). The average length of these transcripts was 1,790 bp with an average of 6.8 exons, which was larger than the size of the lincRNA genes. However, the average exon length of the protein-coding genes was 261 bp, which was less than that of the lincRNA genes. Furthermore, the exon size distribution of protein genes was mostly within 200 bp (Figure 2A). We found that protein transcripts that contained only two exons accounted for 14.4% of the total protein-coding genes, which was far less than what was observed for the lincRNA genes (Figure 2B).

Comparison of features of porcine lincRNAs and protein-coding genes.
Conservation of porcine lincRNAs
To examine the sequence conservation of lincRNAs between pig and other mammals, we compared pig lincRNAs with those of mouse and human lincRNAs (Ensembl Genes 70) using BLASTN version 2.2.26+. The Ensembl database contains 11,325 human lincRNAs and 3,148 mouse lincRNAs. Only 28 (5%) and six (1.05%) pig lincRNAs overlapped with human and mouse lincRNAs (E-value ≤10−5), respectively. Additionally, four of the lincRNAs corresponded to two lincRNA genes overlapping between human and mouse. The lincRNAs conserved between pig, human and mouse spanned a modest portion of the transcript ranging from 28 to 2,282 nt (396 nt on average) and 35 to 1,930 nt (612 nt on average), respectively. We further found that six and three pig lincRNAs had sequence homology with human and mouse lincRNAs, respectively, restricted to the regions located in a single exon in which the conserved regions started from or ended at the intron-exon boundary (Figure 3). In these nine cases, both the pig lincRNA and the mammalian ortholog were also spliced, indicating that the relative position of the exon within the conserved region was conserved.

Representative images of two pig lincRNAs with conserved segments for mouse (A) and human (B).
Thick lines indicate an exon and thin lines indicate an intron of the lincRNA. Boxes indicate the conserved region between the two lincRNAs.
In addition, we found that 364 and 137 porcine lincRNA loci could be synthetically mapped to the human and mouse genome using liftover with a value of 0.5 for the “Minimum ratio of bases that must remap”. Moreover, we found that 91 porcine lincRNA loci overlapped (>1 bp) with the human lincRNA loci, which also contained 19 lincRNAs of the above 28 lincRNAs. In addition, 20 porcine lincRNA loci overlapped (>1 bp) with the mouse lincRNA loci, which contained all of the above six lincRNAs.
Repetitive elements of porcine lincRNA
We found that 367 (65%) pig lincRNA transcripts harbored at least a partial repetitive element (RE) and that 12.64% of the lincRNA transcripts are composed of ≥50% RE-derived sequences (Figure 4). In general, the number of lincRNAs decreased with the increasing RE content ratio (Figure 4). The average size of the RE-derived fragments in the lincRNAs was 355 bp, whereas the average length of the 367 lincRNAs was 1,263 bp. Thus, on average, 28% of the lincRNA length is composed of REs. Short interspersed repetitive sequences (SINEs) and long interspersed repetitive sequences (LINEs) accounted for 57.64% of the total REs and the long terminal repeat (LTR) occupied 15.52% of the total RE. In addition to the above major REs, 19.25% of REs were identified as simple repeat and low complexity sequences.

Percentage of pig lincRNA transcripts masked by RE (from 0 to 100%).
Nearest neighbor analysis of lincRNA genes
We found that 259 lincRNA loci were transcribed near (<10 kb) their protein-coding neighbor and a total of 378 protein-coding neighbors were collected. Of these neighbors, 361 were assigned to 26 GO terms involved in the biological process (Table 1, P < 0.05). The 26 GO terms mainly referred to the development process, transcriptional regulation and the biosynthetic process. We further found that all of the GO terms contained a small number of genes, except those related to transcription regulation and the biosynthetic process.
Expression of lincRNAs at different developmental stages
We randomly selected 10 lincRNAs and examined their expression pattern at three important development stages. The results confirmed the expression of seven lincRNAs, of which six were detected at all time points (Figure 5) and showed differential expression between the fetal and adult periods. We further found that CUFF.15945 and CUFF.6127 were both higher in the 65 dpc period and were considerably decreased during muscle development; they had a distinctive expression pattern compared with the other four lincRNAs.

Developmental expression pattern of lincRNAs during muscle development (here and below, the values represent the means ± s.e.m., n = 6).
Characterization of ENSMUSG00000090086
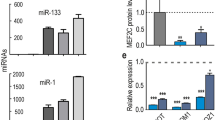
We choose a mouse lincRNA gene locus (named ENSMUSG00000090086) that was conserved between pig and human to analyze its features during C2C12 cell differentiation. The ENSMUSG00000090086 gene contained seven transcript isoforms and we found that iso3 and, in particular, iso1 and iso4 were considerably up-regulated during C2C12 cell differentiation; whereas iso2, iso5, iso6 and iso7 were not detected (Figure 6A). We separated the differentiated C2C12 cells into nuclear and cytoplasmic fractions and found that iso1 and iso4 were both expressed mainly in the nucleus, although iso4 showed weak expression a weak expression (Figure 6C, Supplementary Information). Further, in RNA immunoprecipitation (RIP) experiments, we confirmed that iso1 and iso4 were both significantly (P < 0.01) enriched with EZH2 antibody compared to the IgG nonspecific antibody (Figure 6D).

Features of the ENSMUSG00000090086 gene.
(A) Q-PCR analysis of ENSMUSG00000090086 gene expression during C2C12 cell culture in growth medium (GM) or differentiation medium (DM) for 1, 3 and 5 days (here and below, the values represent the means ± s.e.m., n = 6). (B) RT-PCR analysis of Myog and MHC expression to monitor the differentiation status at indicated times. HPRT was used as an endogenous control. (C) RT-PCR analysis of the relative expression of iso1 and iso4 as well as other control genes in the nuclear and cytoplasmic cell fractions. (D) The RIP result of iso1, iso4 and HOTAIR with the EZH2 antibody. RIP enrichment was measured by q–PCR and the values were normalized to background levels and input samples (Percent Input Method). The interaction of HOTAIR with EZH2 is a known interaction that served as a positive control(**indicates P < 0.01, here and below, the values represent the means ± s.e.m., n = 3).
Discussion
The identification and characterization of porcine lincRNA, particularly in fetal skeletal muscle development, has been very limited compared with that of lincRNAs in humans39,45 and other model organisms, such as zebrafish14,46 and mouse5,35. To the best of our knowledge, this is the first report of the systematic identification and characterization of a reference catalog of 570 porcine lincRNAs by integrating RNA-seq data from fetal muscle tissues. We annotated the basic features of the pig lincRNAs, including the transcript structure, sequence conservation, transposable elements, nearest neighbor analysis and developmental expression. Below, we discuss these findings in more detail and relate our findings to results that have emerged from humans and other model organisms.
Noncoding and protein-coding genes were distinguished by their coding potential capability. It has been reported that CPC can discriminate coding from noncoding transcripts with high accuracy47. Additionally, some reports have shown that the combination of a strict BlastX and Pfam (PfamA and PfamB) search could better reduce false negative and false positive results39,46. Therefore, we performed this combination of steps to ensure that our resultant lincRNAs were of high quality.
We noted that about 73.1% of the total clean reads were mapped to Sus scrofa genome assembly v.10.2, which showed a low efficiency of mapping. Here we only focus on the number of reads that could be mapped to all the 20 chromosomes (SSC1-18, X and Y), and, thus, we observed a low efficiency of mapping. Nevertheless, when the reads were mapped to the 20 chromosomes and unplaced scaffolds, the mapping rate was increased to 80.4%, which was in accordance with previous reports48.
LincRNA genes are typically shorter (~1 kb) and have fewer exons (~2–3) than protein-coding genes39,46 and, thus, have relatively simple compositions. Our putative porcine lincRNAs also display these properties, indicating that the lincRNAs identified here were reliable. However, an earlier study showed that porcine lincRNAs identified in the testis were 456 bp in size on average40, which is approximately half the size of the lincRNAs in human39 and swine characterized here. This inconsistency is possibly due to fewer reads and mapping problems. Our results showed that the porcine lincRNAs had 1.2 isoforms per locus, which was lower than that of human lincRNAs39. This difference does not seem to be attributable to the fewer reads, as zebrafish lincRNAs had more sequence reads compared to human lincRNAs, although they exhibited less efficient alternative splicing39,46. We also found that our lincRNAs had canonical splice sites (GT/AG), which supported the fact that the lincRNAs were similar to protein-coding genes in some properties, such as chromatin modification and splicing signals39,49.
We found that almost half of the porcine lincRNA was transcribed near (<10 kb) protein-coding genes, which is consistent with the finding that lincRNA genes were preferentially found within 10 kb of protein-coding genes2,49. However, some studies showed different results50, which may be attributed to the diverse sources of lincRNA. It was demonstrated that lincRNAs were transcribed in close proximity to protein-coding genes and was possibly coordinated with transcriptional regulation of neighboring coding genes51. This was partially supported by evidence that mammalian lincRNAs (<10 kb) are more likely to be located near genes that mediate transcriptional regulation14,39, which was also observed in the GO analysis in the nearest neighbors of porcine lincRNA genes.
Transposable elements comprise a substantial fraction of the vertebrate genome1 and have been shown to be a major source of vertebrate lncRNAs52. In this study, most of the porcine lincRNAs were also composed of partial TE-derived sequences, which is consistent with the above conclusion. Moreover, we found that SINEs and LINEs account for half of the TE family in the porcine lincRNAs, which is also consistent with the results observed in the mammalian lncRNAs52.
Most of the lincRNAs had a less conserved sequence with other mammalian lincRNAs and some of them had more positional conservation than sequence conservation across vertebrates14. Our results showed that few pig lincRNAs overlapped with the human and mouse lincRNAs at the sequence level, but more pig lincRNAs were synthetically mapped to the mammalian genome, which is consistent with the above conclusions. However, a recent report showed that nearly 40% of pig lincRNAs had detectable sequence homology with human and mouse lincRNAs by BLASTN. We found that our pig lincRNAs were only obtained from the skeletal muscle, while the 6,621 pig lincRNAs in Zhou et al. (2014) were obtained from several tissues53. LincRNAs exhibited tissue-specific expression patterns more so than protein-coding genes, which may lead to identify different number and structure of lincRNAs across multiple tissues. Furthermore, the mouse lincRNAs from the NONCODE database (v4)54 and human lincRNAs from the Gencode database (v19)55 database was also different from the mouse and human lincRNAs database (Ensemble Genes 70) analysed in this study. Thus, we think the difference in the number and source of pig lincRNAs and the databases of mouse and human lincRNAs may have led to our poor results about lincRNAs sequence similarity between species.
Conserved lincRNAs among mammals are generally thought to have important roles14. We found that the conserved ENSMUSG00000090086 gene was differentially expressed during C2C12 cell differentiation and was associated with PRC2. It was shown that the binding of differentially expressed lincRNAs to PRC2 could indicate a possible role of lincRNAs8. However, a recent study stated that the binding to PRC2 does not necessarily imply functionality for lincRNA56. The role of the ENSMUSG00000090086 gene needs to be further investigated.
In conclusion, we have provided a resource of porcine lincRNA which will enable further studies of the function of these genes in the process of skeletal muscle development.
Methods
The methods were performed in accordance with the guidelines of the Good Experimental Practices adopted by the Institute of Animal Science.
All experimental protocols were approved by the Institute of Animal Science of the Chinese Academy of Agricultural Sciences.
Animal and tissue preparation
All longissimus dorsi muscle samples, which were maintained in liquid nitrogen, were derived from our laboratory. Two Tongchen pig fetuses (one male and one female) at 50, 55, 60, 65 and 75 dpc were included in this study.
RNA extraction, library preparation and Solexa sequencing
Total RNA was isolated with TRIzol reagent (Invitrogen, Carlsbad, CA, USA), treated with DNase I (Qiagen, Beijing, China) and purified using an RNeasy MinElute Cleanup column (Qiagen). The total RNA integrity was assessed using Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) and only the samples with RNA Integrity Number (RIN) scores >8 were used for sequencing. Equal amounts of total RNA from the samples at the different stages (i.e., 50, 55, 60, 65 and 75 dpc) were pooled into one sample.
PolyA+ RNA was purified using Magnetic Oligo (dT) Beads from 20 μg of the total RNA and was further fragmented before cDNA synthesis. First-strand cDNA was synthesized using Random Primer p(dN)6 and Superscript III (Invitrogen, Carlsbad, CA, USA) and the synthesis of double-stranded cDNA was performed using 10× second strand buffer, RNaseH and DNA Polymerase I. Following the second-strand cDNA synthesis and adaptor ligation, 240–310 bp cDNA fragments were isolated. The cDNA libraries were then prepared following the manufacturer's instructions (Illumina, San Diego, CA, USA). The purified cDNA libraries were sequenced using a paired-end sequencing strategy on the Illumina HiSeq 2000 after quantification by the Agilent 2100 Bioanalyzer.
Transcriptome assembly
The raw reads were cleaned by filtering the adapter using cutadapt v1.157 and low-quality reads using Prinseq v0.17.358. The clean reads were then mapped to the pig reference genome (Sscrofa10.2) using the TopHat version 1.3.2 software59. Transcriptomes were assembled with Cufflinks version 1.3.038 supported through Galaxy (https://usegalaxy.org/u/jeremy/w/sort-sam-file-for-cufflinks).
Pipeline for the identification of multiple-exon lincRNA
We identified multiple-exon lincRNAs following the steps listed in the pipeline (Figure 1). The steps are detailed as follows:
(1) Size selection: single-exon transcripts and the transcripts less than 200 bp were removed; (2) the remaining transcripts were removed if they had genomic positions that overlapped (>1 bp) with those of pig protein-coding genes obtained from NCBI RefSeq mRNAs (release 54, July 2012) with the accession prefixes NM_ and XM_ (hypothetical protein genes were not included) and Ensembl protein-coding genes (Ensembl release 68, July 2012); (3) the Coding Potential Calculator (CPC) tool47 was used to assess the coding potential of transcripts in both strands and the remaining transcripts were removed if they had a CPC value >0 in either strand; (4) any remaining transcripts with similarity to known proteins against the Swiss-Prot database with an E-value ≤10−5 were removed using the NCBI BLAST version 2.2.26; (5) the remaining transcripts that contained a known protein-coding domain were removed. To accomplish this, we translated each transcript sequence in all six possible frames and used HMMER to exclude the transcripts whose corresponding translated protein sequences had a significant hit in the Pfam (PfamA and PfamB) database release 26.060; (6) the remaining transcripts that belonged to known classes of small RNAs (snRNA, snoRNAs, tRNAs, miRNA, etc.) were removed using Rfam61 (release 10.0); and (7) to filter the transcripts that were located in the UTR regions of the protein-gene due to incomplete assemblies, the remaining sequences were aligned against the NCBI RNA reference sequences (RefSeqs) only with the identifiers beginning with “NM_” prefixes via BLASTN. The sequences with more than 90% of their lengths overlapping in the UTR regions of the RNA RefSeqs were discarded.
Analysis of protein-coding transcript, TEs and GO
The assembled transcripts that had at least two exons were collected and were considered to be protein-coding transcripts when they had a sequence similarity of ≥96% and overlapped with ≥90% of the porcine protein genes from NCBI RefSeq mRNAs (hypothetical protein genes were not included; release 54, July 2012) and Ensemble (release 68, July 2012) using BLASTN version 2.2.26+ (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/).
We ran RepeatMasker program version open-4.0.3 (http://www.repeatmasker.org/) with options “cross_match” as the search engine and “pig” as the DNA source to identify transposable and repetitive DNA elements in the pig lincRNA sequences.
For each lincRNA locus, the nearest upstream and downstream (within <10 kb) protein-coding neighbors (without overlap) were identified. The neighbor gene names were used as the gene list input into DAVID (http://david.abcc.ncifcrf.gov/) for GO analysis62. We selected the “GOTERM_BP_FAT” and set the value of EASE to 0.05 for the GO term enrichment analysis.
Reverse transcription polymerase chain reaction (RT-PCR) and real-time PCR
For RT-PCR, the total RNA was converted into cDNA using a Revert Aid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Boston, MA, USA) with oligo dT and random hexamer primers included in the kit. The PCR reactions were performed as follows: initial denaturation at 95°C for 3 min, followed by 30 cycles of denaturation at 95°C for 15 s, annealing at 60°C for 30 s and elongation at 72°C for 20 s. The real-time PCR was performed according to the SYBR Premix Ex Taq™ instructions (Takara, Shiga, Japan). The reaction volume contained 10 μl of 2× SYBR® Premix Ex Taq™, 0.4 μl of 50× ROX Reference Dye II, 0.5 μl of 10 μM forward and reverse primers, 2 μl of template cDNA and dH2O up to a final volume of 20 μl. The reactions were performed on an ABI 7500 instrument (Applied Biosystems, Carlsbad, CA, USA) as follows: 2 min at 95°C, followed by 35 cycles of 5 s at 95°C and 34 s at 60°C. All data were analyzed by the 2−ΔΔCT method using 7500 System (SDS) Software version 1.4.0. LincRNA primers (Table 2) were designed over suitable exon-exon junctions using primer3 (http://bioinfo.ut.ee/primer3-0.4.0/primer3/input.htm).
Cell culture and differentiation
The mouse C2C12 cell line was provided by the Cell Resource Center of Peking Union Medical College (CRC/PUMC, Beijing, China) and was cultured in Dulbecco's modified Eagle's medium (DMEM) - high glucose (Invitrogen, Carlsbad, CA, USA) with 10% (v/v) fetal bovine serum (Invitrogen, Carlsbad, CA, USA) at 37°C in a 5% CO2 humidified incubator. Upon the induction of differentiation, the culture medium was switched to DMEM plus 2% horse serum when cells reached approximately 80% confluence. The medium was changed every two days.
Nuclear and cytoplasmic RNA fractionation
The cells cultured in differentiation medium were harvested in a T-25 flask, washed once with cold PBS and centrifuged at 500 g for 3 min at 4°C. Cell pellets were resuspended by gentle pipetting in 200 μl of lysis buffer (10 mM Tris-HCl, pH = 8.0, 140 mM NaCl, 1.5 mM MgCl2, 10 mM EDTA, 0.5% IGEPAL® CA-630 and 40 U/ml RNase inhibitor) and incubated on ice for 5 min, followed by centrifugation at 500 g at 4°C for 3 min. The supernatant was transferred to a fresh 1.5 ml microcentrifuge tube, centrifuged at full speed (14,000 rpm) for 1 min and lysed in 1 ml of TRIzol for cytoplasmic RNA isolation. The nuclear pellet was washed once with lysis buffer and was resuspended in 200 μl of lysis buffer to determine the nucleus viscosity; 1 ml of TRIzol was added for nuclear RNA isolation if necessary. An equal amount of nuclear and cytoplasmic RNA was reverse transcribed for further analysis.
RNA-binding protein immunoprecipitation (RIP)
Cells cultured in differentiation medium were prepared in four T-75 flasks for the RNA-binding protein immunoprecipitation (RIP) assay using an EZ-Magna RIP Kit (Millipore, Billerica, MA, USA) and following the manufacturer's instructions. An anti-EZH2 polyclonal antibody (ab3748, 1:100; Abcam, Cambridge, UK) and negative control rabbit IgG antibody was used to investigate the interactions between lincRNAs and PRC2. The final isolated RNA was reverse transcribed using random primers according to the RevertAid™ First-Strand cDNA Synthesis Kit. Data were analyzed using the Percent Input Method (http://www.lifetechnologies.com/cn/zh/home/life-science/epigenetics-noncoding-rna-research/chromatin-remodeling/chromatin-immunoprecipitation-chip/chip-analysis.html).
Statistical analysis
The results are reported as the mean ± standard deviation (SD). Statistical analysis was performed using a two-tailed unpaired Student's t-test. Differences were considered statistically significant at the p < 0.05 level and were considered very significant at p < 0.01. All experiments were performed three times.
References
Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W. & Mural, R. J. The sequence of the human genome. Science 291, 1304–1351 (2001).
Bertone, P. et al. Global identification of human transcribed sequences with genome tiling arrays. Science 306, 2242–2246 (2004).
Birney, E. et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816 (2007).
Mercer, T. R. et al. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat Biotechnol 30, 99–104 (2012).
Guttman, M. et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 458, 223–227 (2009).
Brannan, C. I., Dees, E. C., Ingram, R. S. & Tilghman, S. M. The product of the H19 gene may function as an RNA. Mol Cell Biol 10, 28–36 (1990).
Brown, C. J. et al. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 349, 38–44 (1991).
Khalil, A. M. et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A 106, 11667–11672 (2009).
Ørom, U. A. et al. Long noncoding RNAs with enhancer-like function in human cells. Cell 43, 46–58 (2010).
Penny, G. D., Kay, G. F., Sheardown, S. A., Rastan, S. & Brockdorff, N. Requirement for Xist in X chromosome inactivation. Nature 379, 131–137 (1996).
Lee, J. T. Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev 23, 1831–1842 (2009).
Loewer, S. et al. Large intergenic non-coding RNA-RoR modulates reprogramming of human induced pluripotent stem cells. Nat Genet 42, 1113–1117 (2010).
Guttman, M. et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 477, 295–300 (2011).
Ulitsky, I., Shkumatava, A., Jan, C. H., Sive, H. & Bartel, D. P. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell 147, 1537–1550 (2011).
Clemson, C. M. et al. An Architectural Role for a Nuclear Noncoding RNA: NEAT1 RNA Is Essential for the Structure of Paraspeckles. Mol Cell 33, 717–726 (2009).
Mao, Y. S., Sunwoo, H., Zhang, B. & Spector, D. L. Direct visualization of the co-transcriptional assembly of a nuclear body by noncoding RNAs. Nat Cell Biol 13, 95–101(2011).
Kanis, E., De Greef, K. H., Hiemstra, A. & van Arendonk, J. A. Breeding for societally important traits in pigs. J Anim Sci 83, 948–957 (2005).
Rehfeldt, C., Fiedler, I., Dietl, G. & Ender, K. Myogenesis and postnatal skeletal muscle cell growth as influenced by selection. Livest Prod Sci 66, 177–188 (2000).
Muráni, E., Murániová, M., Ponsuksili, S., Schellander, K. & Wimmers, K. Identification of genes differentially expressed during prenatal development of skeletal muscle in two pig breeds differing in muscularity. BMC Dev Biol 7, 109 (2007).
International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature 431, 931–945 (2004).
Cheng, J. et al. Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308, 1149–1154 (2005).
Birney, E. et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816 (2007).
Kapranov, P. et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316, 1484–1488 (2007).
Wu, S. C., Kallin, E. M. & Zhang, Y. Role of H3K27 methylation in the regulation of lncRNA expression. Cell Res 20, 1109–1116 (2010).
Zhao, S. H. et al. Complementary DNA macroarray analyses of differential gene expression in porcine fetal and postnatal muscle. J Anim Sci 81, 2179–2188 (2003).
Te Pas, M. F. et al. Transcriptome expression profiles in prenatal pigs in relation to myogenesis. J Muscle Res Cell Motil 26, 157–165 (2005).
Tang, Z. et al. LongSAGE analysis of skeletal muscle at three prenatal stages in Tongcheng and Landrace pigs. Genome Biol 8, R115 (2007).
Zhao, X. et al. Comparative analyses by sequencing of transcriptomes during skeletal muscle development between pig breeds differing in muscle growth rate and fatness. PloS one 6, e19774 (2011).
Huang, T. H., Zhu, M. J., Li, X. Y. & Zhao, S. H. Discovery of porcine microRNAs and profiling from skeletal muscle tissues during development. PloS one 3, e3225 (2008).
Nielsen, M. et al. MicroRNA identity and abundance in porcine skeletal muscles determined by deep sequencing. Anim Genet 41, 159–168 (2010).
Zhou, B., Liu, H. L., Shi, F. X. & Wang, J. Y. MicroRNA expression profiles of porcine skeletal muscle. Anim Genet 41, 499–508 (2010).
Hou, X. et al. Discovery of MicroRNAs associated with myogenesis by deep sequencing of serial developmental skeletal muscles in pigs. PloS one 7, e52123 (2012).
Caretti, G., Di Padova, M., Micales, B., Lyons, G. E. & Sartorelli, V. The Polycomb Ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev 18, 2627–2638 (2004).
Khalil, A. M. et al. Many human large intergenic noncoding RNAs associate with chromatin-modifying complexes and affect gene expression. Proc Natl Acad Sci U S A 106, 11667–11672 (2009).
Guttman, M. et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat Biotechnol 28, 503–510 (2010).
Xiao, B. et al. Identification, bioinformatic analysis and expression profiling of candidate mRNA-like non-coding RNAs in Sus scrofa. J Genet Genomics 36, 695–702 (2009).
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L. & Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5, 621–628 (2008).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28, 511–515 (2010).
Cabili, M. N. et al. Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev 25, 1915–1927 (2011).
Esteve-Codina, A. et al. Exploring the gonad transcriptome of two extreme male pigs with RNA-seq. BMC Genomics 12, 552 (2011).
Li, T. et al. Identification of long non-protein coding RNAs in chicken skeletal muscle using next generation sequencing. Genomics 99, 292–298 (2012).
Wigmore, P. M. & Stickland, N. C. Muscle development in large and small pig fetuses. J Anat 137, 235–245 (1983).
Ashmore, C. R., Addis, P. B. & Doerr, L. Development of muscle fibers in the fetal pig. J Anim Sci 36, 1088–1093 (1973).
Swatland, H. J. Muscle growth in the fetal and neonatal pig. J Anim Sci 37, 536–545 (1973).
Derrien, T. et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution and expression. Genome Res 22, 1775–1789 (2012).
Pauli, A. et al. Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res 22, 577–591(2012).
Kong, L. et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res 35, W345–349 (2007).
Corominas, J. et al. Analysis of porcine adipose tissue transcriptome reveals differences in de novo fatty acid synthesis in pigs with divergent muscle fatty acid composition. BMC Genomics 14, 843 (2013).
Ponjavic, J., Ponting, C. P. & Lunter, G. Functionality or transcriptional noise? Evidence for selection within long noncoding RNAs. Genome Res 17, 556–565 (2007).
Jia, H. et al. Genome-wide computational identification and manual annotation of human long noncoding RNA genes. RNA 16, 1478–1487 (2010).
Ponting, C. P., Oliver, P. L. & Reik, W. Evolution and functions of long noncoding RNAs. Cell 136, 629–641(2009).
Kapusta, A. et al. Transposable elements are major contributors to the origin, diversification and regulation of vertebrate long noncoding RNAs. PLoS Genet 9, e1003470 (2013).
Zhou, Z. Y. et al. Genome-wide identification of long intergenic noncoding RNA genes and their potential association with domestication in pigs. Genome Biol Evol 6, 1387–1392 (2014).
Xie, C. et al. NONCODEv4: exploring the world of long non-coding RNA genes. Nucleic Acids Res 42, D98–D103 (2014).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22, 1760–1774 (2012).
Kaneko, S., Son, J., Shen, S. S., Reinberg, D. & Bonasio, R. PRC2 binds active promoters and contacts nascent RNAs in embryonic stem cells. Nat Struct Mol Biol 20, 1258–64(2013).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17, 10–12 (2011).
Schmieder, R. & Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864 (2011).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111(2009).
Bateman, A. et al. The Pfam protein families database. Nucleic Acids Res 30, 276–280 (2002).
Griffiths-Jones, S., Bateman, A., Marshall, M., Khanna, A. & Eddy, S. R. Rfam: an RNA family database. Nucleic Acids Res 31, 439–441(2003).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat Protoc 4, 44–57 (2009).
Acknowledgements
This study was supported by grants from National Nature Science Foundation of China (Nos. 31330074 and 31172189), National High Technology Research and Development Program of China (No. 2012AA020603), National Key Basic Research Program of China (2015CB943100) and Agricultural Science and Technology Innovation Program (ASTIP-IAS05) of Chinese Academy of Agricultural Sciences (CAAS).
Author information
Authors and Affiliations
Contributions
K.L. conceived and designed the experiments. W.Z. and Y.M. performed the experiments. L.M., C.W. and X.H. analyzed the data. Z.T., S.Y., R.Z. and X.H. helped interpret the results. W.Z. and M.-H.L. wrote the paper. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhao, W., Mu, Y., Ma, L. et al. Systematic identification and characterization of long intergenic non-coding RNAs in fetal porcine skeletal muscle development. Sci Rep 5, 8957 (2015). https://doi.org/10.1038/srep08957
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep08957
This article is cited by
-
Transcriptome and methylome sequencing reveals altered long non-coding RNA genes expression and their aberrant DNA methylation in equine sarcoids
Functional & Integrative Genomics (2023)
-
Mammary epithelial cell transcriptome reveals potential roles of lncRNAs in regulating milk synthesis pathways in Jersey and Kashmiri cattle
BMC Genomics (2022)
-
Comprehensive analysis of lncRNAs involved in skeletal muscle development in ZBED6-knockout Bama pigs
BMC Genomics (2021)
-
Analysis of long intergenic non-coding RNAs transcriptomic profiling in skeletal muscle growth during porcine embryonic development
Scientific Reports (2021)
-
Analysis of long non-coding RNAs in skeletal muscle of Bama Xiang pigs in response to heat stress
Tropical Animal Health and Production (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
