
Abstract
A human cell surface displays many complex-structured receptors for receiving extracellular signals to regulate cellular functions. The use of precisely regulated signal-controls of the receptors could have possibilities beyond the current synthetic biology research that begins with the transfection of exogenous molecules to rewire intracellular circuits. However, by using a current ligand-receptor technique, the configuration of the artificially assembled cell surface molecules has been undefined because the assemblage is an unsystematic molecular clustering. Thus, the system bears improvements for precisely regulating receptor functions. We report here a new tool that refines stereochemically-controlled positioning of an assembled surface receptor. The tool performs rationally as an ON/OFF switch and is finely tunable so that a 3 to 6 nm size difference of the device precisely distinguishes the efficiency of apoptosis induced via cell-surface receptor binding. We discuss the potential use of the device in next-generation synthetic biology and in cell surface studies.
Similar content being viewed by others
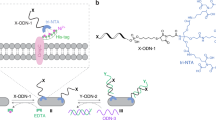


Introduction
Engineered genetic circuits have been developed to establish simple and independent control of biological processes in synthetic biology. Lately, artificial regulations of signal transduction pathways have been reported to control cell fate and to build biocomputing systems1. In either case, the process of synthetically engaging the signal pathways begins with the transfection of molecules such as plasmids, RNA or antibiotics. Meanwhile, cell-surface receptors are activated to turn on the signal transduction cascades with ligand binding, indicating that artificially activated receptors can be used as triggers for regulating original or re-wired channels to perform desired tasks. Thus, the use of synthetic signal controllers of receptors could have research possibilities beyond intracellular synthetic biology.
For artificial activation of the cell surface receptors, the receptor clustering has been investigated with nanoscale synthetic ligands2. In its most advanced form, multiple ligands are attached to a soluble biopolymer to yield multivalent conjugates3. However, the relative configuration of the multivalent ligands is undefined and obscure because they are attached to a freely mobile biopolymer whose behavior is highly elastic by nature. Therefore, the polymer system is unsuitable for investigating the receptor complex assembly (specifically, the inter-receptor distance, the relative configulation and the orientation of the receptors) and other atypical cell-surface receptors that require a sophisticated structurally-ordered assembly4.
For example, a densely packed assembly is essential for the distance-dependent molecular interactions of cell-surface receptors such as TNFR5, glycoprotein receptors6 and growth factor receptors7. These interactions regulate mammalian cellular functions including apoptosis, signal transduction, enzymatic reactions and cell-cell interactions8. The receptor molecules must be precisely assembled to enhance the molecular interactions required for initiating signal transduction9. Likewise, the polymer system bears improvements for precisely regulating the cell-surface receptor function in synthetic biology.
Nucleic acids have been used as tools on the cell surface in bionanotechnology. For example, DNA nanoarrays based on antibody-cell surface interactions have been explored for directing cell-surface interactions to position multiple cells onto a DNA fabric10. An RNA particle containing folate has also been reported to specifically bind to tumor cells11. Moreover, peptide nucleic acids have been exploited as scaffolds to conjugate self-assembled antibody multimers and induce cytotoxic activity12. However, the effectiveness of the RNA-protein complexes (RNPs), particularly their spatial structure, to regulate physiological functions has been unexplored. A designed RNP containing properly deployed binding proteins for a target receptor is conceivably advantageous for the avidity-based alignment of receptors with defined intermolecular distances on their cell surfaces.
We report here a new RNP tool that refines stereochemically-controlled positioning of an assembled receptor on a cell surface to advance the cell-surface analysis. The RNP serves as an unprecedented device for ON/OFF switches and fine-tuning of cell-surface receptors to control apoptosis of a human cell. The RNP with a designable size and shape at nano level can be customized as a high-precision nano-device integrated into re-wired signaling networks in synthetic biology.
Results
Design and construction of a series of equilateral-triangular RNPs
First, we designed RNPs composed of a series of equilateral-triangular RNAs containing the boxC/D sequence, a protein binding RNA motif for the L7Ae protein, in its three apexes in silico13,14. The length of the three sides of the RNA was altered systematically to produce a series of triangular RNPs that insulated the attached proteins via a robust double-stranded stem RNA. Five equilateral Tri-RNAs having 15, 26, 48, 70 and 92 bp (14, 17, 23, 29 and 34 nm by length, respectively) on each side were prepared and termed Tri-RNA15, Tri-RNA26, Tri-RNA48, Tri-RNA70 and Tri-RNA92, respectively (Fig. 1). Electrophoretic mobility shift assays (EMSA) showed that the three binding motifs in the Tri-RNAs interacted with L7Ae to produce a corresponding shifted band (Supplementary Fig. 1a, filled arrowheads). Most of the band corresponding to the free RNA was shifted in the presence of 4.5 times more amount of L7Ae. The estimated dissociation constant between L7Ae and the box C/D motif was between 150 and 300 nM. The RNPs were visualized in liquid with HS-AFM (Fig. 1, Supplementary Fig. 1b, c). Small attachments recognized as L7Ae were observed at the three apexes of the triangular objects (Fig. 1, yellow arrowheads), indicating that each Tri-RNA possessed three bound L7Ae proteins as designed. The assembly yield based on the AFM images looks lower than that based on the EMSA because the AFM imaging was performed in water without salt and pH adjustment to avoid unacceptable noise signals to the images.

Illustration of Tri-RNPs used in this study and their HS-AFM images.
A series of the triangular RNPs (Tri-RNA-L7Ae complexes) with the different side lengths is displayed clockwise on white and grey rectangular tiles (left). The numbers noted represent the number of the base pairs in each side of the Tri-RNA. The length of each Tri-RNA-L7Ae complex (Tri-RNP) is 14, 17, 23, 29 and 34 nm for Tri-RNP15, 26, 48, 70 and 92, respectively. The widths of the rectangles are 10 nm. A purple object in the 3D models represents the L7Ae protein. A blue and green represent the L- and S-strand of the RNAs, respectively. A red is denoted as a boxC sequence in the L-strand and a yellow as a boxD sequence in the S strand. Each Tri-RNP is assessed in HS-AFM (right). The L7Ae proteins in Tri-RNA26 are pointed with yellow arrowheads. A scale bar represents 20 nm.
Verification of RNP binding
Next, novel Tri-RNPs were prepared to contain multiple antibodies at the apexes. A fusion protein L7Ae-Gβ1x2, consisting of L7Ae and two antibody-binding domains in streptococcal protein G (Gβ1), was constructed to fix antibodies at the RNA apexes by sandwiching the Fc domain of an antibody from both sides15. The prospective binding activities of the purified L7Ae-Gβ1x2 to the RNA and anti-CD61 antibody were confirmed by EMSA (Supplementary Fig. 2a) and western blotting analyses (Supplementary Fig. 2b). The extent of gel-mobility shift of the RNAs was almost the same to that with L7Ae and the RNAs (Supplementary Fig. 1a), indicating that the binding affinity of the L7Ae Gβ1x2 is comparable to that of L7Ae by itself. As anticipated, the HS-AFM images indicated the presence of the triangular RNA with Gβ1 next to L7Ae at the three apexes (Fig. 2a, left, open arrowheads) in addition to the antibody (Fig. 2a, right, arrowhead). We next examined the avidity of the RNA-L7Ae-Gβ1x2 complexes to the cell surfaces stably expressing CD61. As expected from the in vitro studies, the RNPs, but not the control IgG (Fig. 2b), attached to the cell surface more effectively than the defective mutants (Supplementary Fig. 2c), indicating that the three proteins on the RNA could augment adherence to the cell surface receptors.

Binding of Tri-RNA-L7Ae-Gβ1x2-anti-CD61 antibody complexes to the cell surface.
(a), The Tri-RNP48 complexes were visualized in the presence (right) or the absence (left) of the anti-CD61 antibody (purple) by HS-AFM. The objects marked by the open and filled arrowheads indicate the Gβ1 domains and antibodies, respectively. The 3D models of the observed complexes are shown under the HS-AFM images. Scale bars represent 50 nm. (b), The 293 cells stably expressing the CD61 antigen are incubated with the anti-CD61 antibodies (top) or control IgG (bottom) followed by wash, Tri-RNP (Tri-RNA-L7Ae-Gβ1x2 prepared by mixing the 10 nM of the RNA and 30 nM of the protein) treatment and the fluorescent microscopic analysis. A red and green indicate the Tri-RNA modified by Alexa 647 and the Azami Green fluorescent protein, respectively. The control IgG is a mouse IgG2a provided by MBL that does not bind the CD61 antigen. A scale bar represents 200 μm.
Size-dependent apoptosis control with designed RNPs
Dimerized galectin-1 (Gal1) promotes the signal transduction for the apoptosis of T cells via an assemblage of glycoreceptors that results in a lattice structure (Fig. 3a)6,16,17,18,19. This effect indicates that multiple Gal1s attached to the Tri-RNA can initiate signal transduction more effectively because they can steadily hold the receptors closer to one another on the cell surface (Fig. 3a)20,21,22. To assess our hypothesis, we designed and constructed a series of Tri-RNAs with L7Ae fused to N-terminally trimmed Gal1 (Gal1β) and that lacked the amino acids for dimerization and pro-apoptotic activity to induce galectin-dependent apoptosis23,24. The binding of L7Ae-Gal1β to the RNA was confirmed by EMSA (Supplementary Fig. 3a). The EMSA indicated that the binding affinity of L7Ae-Gal1β to RNA was comparable to that of L7Ae. The HS-AFM image of a triangular complex with the fusion proteins at the apexes is consistent with the predicted structure of the Tri-RNA with L7Ae-Gal1β (Tri-RNP-Gal1β) (Fig. 3b left). The time-lapse images of the dissociation of L7Ae with another sphere (Fig. 3b right) from the RNA indicated that the sphere was Gal1β at the apexes.

Pro-apoptotic activities of Tri-RNA-L7Ae-Gal1β.
(a), Illustration of apoptosis regulation by manipulating the distances between the receptor components with small and large Tri-RNPs. The receptor components are distributed over the cell surface in the absence of the Tri-RNPs (upper). In the presence of the Tri-RNPs, Gal1βs at each apex in the Tri-RNPs bind to the receptor components and form lattice structures. Small Tri-RNPs gather the receptor components and form the closely packed lattice structure, resulting in apoptosis (lower left). Large Tri-RNPs adversely form the widely spaced lattice structure and inhibit the apoptosis signal (lower right). (b), The HS-AFM image of Tri-RNP48-Gal1β (left) and time lapse images capturing the separation of the L7Ae-Gal1β fusion protein from Tri-RNA48 (right). L7Ae and Gal1β domains are pointed by the filled purple and green arrowheads, respectively. Scale bars represent 50 nm. (c), Fluorescent microscopic images of the Jurkat cells after 30 min incubation with 600 nM of the indicated Tri-RNP and Tri-RNA. RNA is stained with Propidium Iodide (PI) in red and DNA with Hoechst 33342 in blue. A scale bar represents 20 μm. (d), The panel c is quantified and graphed for the indicated Tri-RNPs. The PI intensities are measured in areas 14 pixels within the nuclear envelope excluding the nuclei. The background intensities were subtracted by rolling ball background subtraction algorithm. The error bars are the standard deviation obtained from analyzing 100-3,000 cells. (e), Flowcytometric analysis of the Jurkat cells after 6 hours of incubation with 600 nM of the indicated Tri-RNPs. The cells were stained with annexin-V conjugated with Pacific Blue for the apoptotic cells. The distribution of the cells was graphed according to the Pacific Blue intensities. The cell population between two broken lines was defined as dead cells. The graph represents one of the three independent experiments. (f), The panel e was quantified and graphed for the indicated Tri-RNPs and controls. The error bars are the standard deviation obtained from the three independent experiments.
Subsequently, the series of Tri-RNP-Gal1β constructs of differing sizes (Fig. 1a) were examined for their ability to bind to the cell. The RNPs were mixed with the cells to quantitatively determine their presence on the cell surfaces (Supplementary Fig. 3b). The binding of the RNPs onto the cell surfaces was visualized by fluorescence microscopy (Fig. 3c). The binding states of the RNPs were comparable with a nearly two-fold difference at 600 nM (Fig. 3d, Supplementary Fig. 3c), suggesting the possible formation of lattice structures with the receptors. As expected, the RNPs with the three L7Ae binding sites adhered to the cell much more firmly than those missing one or two sites.
The effects of the RNP series on apoptosis were examined. The cells analyzed by flow cytometry revealed that Tri-RNP15-Gal1β, -26-Gal1β and -48-Gal1β promoted apoptosis and that the efficiency decreased as the size of the Tri-RNA increased (Fig. 3e, f). With respect to the efficiency, L7Ae-Gal1β in the absence of Tri-RNAs exhibited hardly any effect on the apoptosis (Fig. 3f L7Ae-Galβ). In addition, ten times more wildtype Gal1 has been reportedly required to achieve an apoptotic effect comparable with that achieved by Tri-RNP15-Gal1β21. However, the larger RNPs, Tri-RNP70-Gal1β and -92-Gal1β, did not exhibit pro-apoptotic activity. This lack of activity was further confirmed by varying the amount of Tri-RNP15-Gal1β and -92-Gal1β (Supplementary Fig. 3c, d). The progression of a nuclear shrinkage was observed with Tri-RNP15-Gal1β but not with Tri-RNP92-Gal1β (Supplementary Fig. 4a, b). Lactose served as the inhibitor, strongly indicating that the binding of Gal1β triggered apoptosis (Supplementary Fig. 4c)25. Thus, a 3 to 6 nm size difference on one side of the RNP was concluded to precisely determine the efficiency of the signal transduction initiation. The calculated length of each side of Tri-RNP15, 26, 48, 70 and 92 is 14, 17, 23, 29 and 34 nm, respectively (Fig. 1a). The designed smaller RNPs conceivably function as ON switches via a receptor-assemblage, which in return determines the switching efficiency based on how closely packed the structure of the designed RNP is. The designed RNPs that have more closely packed Gal1β molecules at the apexes are more efficient at signaling. This is likely the reason why Tri-RNP15-Gal1β is the most effective.
Insulation of receptor molecules for blocking apoptosis
Adversely, the binding of the larger RNPs, such as Tri-RNP70-Gal1β and -92-Gal1β, could insulate the receptor molecules from one another and prevent the neighbor effect from triggering signal transduction (Fig. 3a, right). This phenomenon implies that larger RNPs can function as OFF switches or limiters. To test this hypothesis, a competition experiment employing Tri-RNA92 and L7Ae-Gal1, was conducted under conditions where L7Ae-Gal1 promotes apoptosis in the presence of Tri-RNA26m3, which lacks the L7Ae binding sites26. As anticipated, apoptosis was blocked competitively in the presence of Tri-RNA92 but enhanced in the presence of Tri-RNA15 (Fig. 4a). Another competitive experiment was successfully performed with Tri-RNP15-Gal1β employing Tri-RNP92-Gal1β as a competitor (Fig. 4b, c). Thus, Tri-RNA92 and Tri-RNP92-Gal1β served as an OFF switch and a limiter, respectively (Fig. 4a, c). This approach for spatially preventing particular molecules from communicating on the cell surface can be useful for specifically inhibiting targeted cellular events in both experimental molecular biology and medicine.

Inhibition of apoptosis by the large Tri-RNP.
(a), The Jurkat cells were mixed with the L7Ae-Gal1 in the presence of the indicated Tri-RNAs. A buffer control does not contain L7Ae-Gal1 or RNA. Following the 6 hours of incubation, the dead cells are counted by flowcytometry and bar-graphed. The error bars represent the standard deviation obtained from three independent experiments. (b), Following 6 hours of incubation with Tri-RNA15-L7Ae-Gal1β at the indicated concentrations in the presence (+) or absence (−) of 600 nM of Tri-RNA92-L7Ae-Gal1β, the Jurkat cells were stained with annexin-V and counted by flowcytometry. The distribution of the cells was graphed according to the Pacific Blue intensities. The cell population between two broken lines was defined as dead cells. The graph represents one of two independent experiments. (c), The panel b is quantified and bar-graphed. The open and filled bars indicate the presence and absence of the 600 nM of Tri-RNP92-L7Ae-Gal1β, respectively. The error bars are the standard deviation obtained from two or more independent experiments.
Discussion
The results illustrate that the molecular design at a nanomolecular level is effective for specifically regulating the functions of the cell-surface receptor. As a next step, the integration of other synthetic control system such as a chimeric receptor with intracellular control devices will greatly enhances the current synthetic ligand-receptor activation system. The intracellular domain of the chimeric receptor enables the precise targeting of a downstream molecule whereas the extracellular domain provides the targeting of a specific ligand partner27,28. Our RNP-based ligand device could provide more sophisticated controls of the receptor assembly to activate or attenuate the signal initiation. This artificial receptor-ligand system has a potential to significantly minimize the undesired crosstalks with other signaling molecules and function as an orthogonal switch which may eventually extend to a more complex extra- and intracellular network in synthetic biology29. Furthermore, since our device allows to tune the signal strength based on the distance between the receptors, it is anticipated that expansion of our device by combining with an internal control system will serve as the independent model to study and rewire the complex cell signaling network30.
We anticipate that this class of nanotechnology provides a feasible and rational approach for manipulating a variety of functional molecules on the human cell surface. The advanced triangular (or other polygonal) RNAs with multiple heterogeneous ligands at the tips enhance or block the assemblage of a variety of heterogeneous cell surface molecules with high precision on a single cell or inter-cellularly. Specifically, the RNP has the potential to block the assemblage of not only receptors but also other cell surface molecules to induce or prevent targeted cellular events with high degree of accuracy.
The design and use of RNPs opens a new route toward the rational design of bio-nanotechnology devices for the mammalian cell surface. The device controls specific cellular events by regulating the chemical affinity, binding specificity, physical organization and signaling control of target molecules. In addition, the materials including DNA, PNA and a number of natural and artificial organic (and inorganic) molecules with specific structures and functions can be incorporated easily to artificially evolve the device.
Methods
RNA preparation
All DNA templates and primers used for transcription were purchased from Hokkaido System Science and Gene Design. For Tri-RNA15, a single stranded template was annealed with the T7 primer and transcribed with MEGAShortscriptTM T7 kit (life technologies) and home-made T7 RNA polymerase. The templates for other longer RNAs were prepared by elongation of hybridized oligonucleotides or PCR with KOD polymerase (TOYOBO) and transcribed as described above. All the transcribed RNAs were gel-purified in denaturing polyacrylamide gels (PAGE). Purified RNAs were dissolved in pure water and stored at −20°C. Concentrations of RNAs and DNAs were determined by ND-1000 (Thermo scientific). Alexa 647-labeled RNAs are made by UlysisTM Alexa Fluor® 647 Nucleic Acid Labeling Kit (Invitrogen) according to the manufacturer's protocols.
T7 primer 5′-GCTAATACGACTCACTATA-3′
Tri-DNA15L 5′-GGTGGAATGCTCATCACGCCCAGGGCCGGCACAGGGCTCATCACGCCGAAGCCGGACTTCTCGCTCATCACGCCCTTCACCTATAGTGAGTCGTATTAGC-3′
Tri-DNA15S 5′-GGCTTCGGTCAGAGCCCTGTGCCGGCCCTGGGTCAGAGCATTCCACCGGTGAAGGGTCAGAGCGAGAAGTCCTATAGTGAGTCGTATTAGC-3′
Tri-DNA26L-fwd 5′-CTAATACGACTCACTATAGGCGCAAAGGCCTGTAATCGGCGTGATG-3′
Tri-DNA26L-rev 5′-TTGCTACGCTCATCACGCCCGGATGGGCATGCTCTTCCCGTAGAGGCTC-3′
Tri-DNA26L-tmp 5′-GGCCTGTAATCGGCGTGATGAGCCATGCGAGGAGGAAATGAAGTCCAATGGCGTGATGAGCCTCTACGGGAAGAGC-3′
Tri-DNA26S-fwd 5′-CTAATACGACTCACTATAGGCCTTTGCGCCTTGCTACG-3′
Tri-DNA26S-rev 5′-TGTAATCGGTCAGAGCCATGCGAGGAGGAAATGAAGTCCAATGGTCAGAG-3′
Tri-DNA26S-tmp 5′-GCCTTTGCGCCTTGCTACGCTCTGACCCGGATGGGCATGCTCTTCCCGTAGAGGCTCTGACCATTGGACTTC-3′
Tri-DNA48L-fwd 5′-CTAATACGACTCACTATAGGACGAGCTGTACACCATGGTGACCGCCGCCGGGCGTGATGAGCTCCAAGGACCCCAACGAGAAGCGCGATCACATGATCTACTTCGGCTT-3′
Tri-DNA48L-rev 5′-GCGCTCTGGGTGGAATGCTCATCACGCCCAGGGCCGTGGGTGATGGCAGGTAGTGGTTATCGGGCAGCAGCACAGGGCTCATCACGCCGAAGCCGAAGTAGATCATGTG-3′
Tri-DNA48S-fwd 5′-CTAATACGACTCACTATAGGTGTACAGCTCGTCCGCGCTCTGGGTGGAATGCTCTGACCCAGGGCCGTGGGTGATGGCAGGTAGTGGTTATCGGGCAGCAGCACA-3′
Tri-DNA48S-rev 5′-ATGGTGACCGCCGCCGGGTCAGAGCTCCAAGGACCCCAACGAGAAGCGCGATCACATGATCTACTTCGGCTTCGGTCAGAGCCCTGTGCTGCTGCCCGATAACC-3′
Tri-DNA70L-1 5′-GCTAATACGACTCACTATAGGACGAGCTGTACACCACACGGACATGACCACCAAAACTCTGGCGTGATGAGCTCTTTTAC-3′
Tri-DNA70L-2 5′-CGTGATGAGCTCTTTTACGGTGCTGATGGGGAGCAGGTCTGACCCGCACGCACGCGGAGGGACCGCAGGCCCTTTCAGGA-3′
Tri-DNA70L-3 5′-GTGATTACATGGCAGCCGCCCGCTTATATTGGTACCTATCGCCACGCTTGGCTCATCACGCCTCCTGAAAGGGCCTGCGG-3′
Tri-DNA70L-4 5′-GCACCGCGAAACCTTGACGTTGACCTCAAGCTCATCACGCCAAACCGCAAATTATCCGTTGGTGATTACATGGCAGCCG-3′
Tri-DNA70S-1 5′-GCTAATACGACTCACTATAGGTGTACAGCTCGTCCGCACCGCGAAACCTTGACGTTGACCTCAAGCTCTGACCAAACC-3′
Tri-DNA70S-2 5′-GTGGCGATAGGTACCAATATAAGCGGGCGGCTGCCATGTAATCACCAACGGATAATTTGCGGTTTGGTCAGAGCTTGA-3′
Tri-DNA70S-3 5′-ATTGGTACCTATCGCCACGCTTGGCTCTGACCTCCTGAAAGGGCCTGCGGTCCCTCCGCGTGCGTGCGGGTCAGACCT-3′
Tri-DNA70S-4 5′-ACACGGACATGACCACCAAAACTCTGGTCAGAGCTCTTTTACGGTGCTGATGGGGAGCAGGTCTGACCCGCACGCA-3′
Tri-DNA92L-1 5′-GCTAATACGACTCACTATAGGACGAGCTGTACACCACACGGACATGACCCAACCAATTACACCAAAACTCTGGCGTGATGAGCTCTTTTACGG-3′
Tri-DNA92L-2 5′-TGAAAGGGCCTGCGGTCCCTCCGCGTGCGTGCGGGTCAGACCGGATACGCAGCATCGGCACACCTGCTCCCCATCAGCACCGTAAAAGAGCTCATCA-3′
Tri-DNA92L-3 5′-GGACCGCAGGCCCTTTCAGGAGGCGTGATGAGCCAAGCGTGGCGATAGGTACCAATCCAAGCCTAATGGTTGCGGTCGATAAGCGGGCGGCTGCCAT-3′
Tri-DNA92L-4 5′-GCACCGCGAAACCTTGAGCGAACTTCCTCGTTGACCTCAAGCTCATCACGCCAAACCGCAAATTATCCGTTGGTGATTACATGGCAGCCGCCCGCTT-3′
Tri-DNA92S-1 5′-GCTAATACGACTCACTATAGGTGTACAGCTCGTCCGCACCGCGAAACCTTGAGCGAACTTCCTCGTTGACCTCAAGCTCTGACCAAACCGCAAAT-3′
Tri-DNA92S-2 5′-AGCGTGGCGATAGGTACCAATCCAAGCCTAATGGTTGCGGTCGATAAGCGGGCGGCTGCCATGTAATCACCAACGGATAATTTGCGGTTTGGTCAGA-3′
Tri-DNA92S-3 5′-GGTACCTATCGCCACGCTTGGCTCTGACCTCCTGAAAGGGCCTGCGGTCCCTCCGCGTGCGTGCGGGTCAGACCGGATACGCAGCATCGGCACAC-3′
Tri-DNA92S-4 5′-ACACGGACATGACCCAACCAATTACACCAAAACTCTGGTCAGAGCTCTTTTACGGTGCTGATGGGGAGCAGGTGTGCCGATGCTGCGTA-3′
Tri-DNA26m1L-fwd 5′- CTAATACGACTCACTATAGGCGCAAAGGCCTGTAATCGGCGTGATGAGCCATGCGAGGAGGAAATGAAGTCC-3′
Tri-DNA26m1L-rev 5′- TTGCTACGCTCATGACGCCCGGATGGGCATGCTCTTCCCGTAGAGGCTCATCACGCCATTGGACTTCATTTCCTCCTCGC-3′
Tri-DNA26m3L-fwd 5′- CTAATACGACTCACTATAGGCGCAAAGGCCTGTAATCGGCGTCATGAGCCATGCGAGGAGGAAATGAAGTCC-3′
Tri-DNA26m3L-rev 5′- TTGCTACGCTCATGACGCCCGGATGGGCATGCTCTTCCCGTAGAGGCTCATGACGCCATTGGACTTCATTTCCTCCTCGC-3′
Tri-DNA26m1S-rev 5′- TGTAATCGGTCAGAGCCATGCGAGGAGGAAATGAAGTCCAATGGTCAGAGCCTCTACGGGAAGAGCATGC-3′
Tri-DNA26m2S-rev 5′- TGTAATCGGGCAGAGCCATGCGAGGAGGAAATGAAGTCCAATGGTCAGAGCCTCTACGGGAAGAGCATGC-3′
Tri-DNA26m3S-fwd 5′- CTAATACGACTCACTATAGGCCTTTGCGCCTTGCTACGCTCTGCCCCGGATGGGCATGCTCTTCCCGTAGAGG-3′
Tri-DNA26m3S-rev 5′- TGTAATCGGGCAGAGCCATGCGAGGAGGAAATGAAGTCCAATGGGCAGAGCCTCTACGGGAAGAGCATGC-3′
Electrophoretic gel-mobility shift assay (EMSA)
The L strands and S strands of RNAs were mixed and incubated at 80°C for 5 min in L7Ae buffer (20 mM HEPES-KOH (pH 7.4), 150 mM KCl, 1.5 mM MgCl2) followed by the incubation at room temperature for 10 min. The annealed RNA (100 nM) was mixed with the indicated concentrations of the proteins. After the incubation at room temperature for 30 min, the RNA products with a loading dye were loaded onto acrylamide gel with 0.5 ×TBE buffer at room temperature. After electrophoresis, the gels were stained with SYBR® Green I/II (Lonza) and analyzed by FLA7000 (GE Healthcare).
Expression and purification of recombinant proteins
The total RNA of HeLa cells was purified by RNAqueous®-4PCR kit (Roche). Reverse transcription of the total RNA was performed by High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The galectin-1 open reading frames (Gal1 and Gal1β) were amplified by PCR using the primers 5′-TATGAGATCTGTCAAAGGCCACACATTTG-3′ and 5′-GACACCATGGGATCCGCTTGTGGTCTGGTCGCCAG-3′ for Gal1 (and 5′-GACACCATGGGATCCAGCAACCTGAATCTCAAACC-3′ for Gal1β). The PCR products were digested by NcoI and BglII and inserted into the cloning vector. After validation of the sequences of the genes, the digested fragments were introduced into the downstream to the L7Ae gene to generate the pET28b(+) expression vectors for the fusion proteins, L7Ae-Gal1 and L7Ae-Gal1β. Both of them contain the amino acids linker, R(SGG)8RS, between L7Ae and galectin-1. One-STrEP-tag and Histidine-tag are located at the N- and C terminus of the expressed proteins, respectively. These expression vectors were transformed into the Escherichia coli strain BL21(DE3). Protein expression was induced at an OD660 of 0.6–0.9 by the addition of IPTG at the final concentration of 1 mM. After 8 hours of incubation at 30°C with shaking, the cells were harvested. The pellets were sonicated and the lysates were centrifuged at 20,000 × g for 30 min at 4°C to remove bacterial debris. The supernatant was passed through a 0.22 μm filter and loaded on 1 mL HisTrap Nickel affinity column by AKTA explorer 10S (GE Healthcare). After washing step, a pulse injection of 150–200 mM of NaOH into the column was performed to remove RNAs non-specifically bound to the proteins and the column. The bound protein was eluted off of the column by the gradient concentrations of imidazole and the fraction enriched in the target protein was determined by SDS-PAGE and Coomassie Brilliant Blue (CBB) staining. Purified proteins were concentrated and dialyzed against the storage buffer (50% glycerol, 14 mM of β-mercaptoethanol in PBS) and the concentration was determined by the absorbance at 280 nm. The proteins were stored at −20°C until used.
High Speed-Atomic force microscopy (HS-AFM)
The RNA was incubated at 80°C for 5 min and then at room temperature for 10 min in L7Ae buffer. The indicated RNA and proteins were mixed at 25 and 250 nM in L7Ae buffer, respectively and incubated at room temperature for 10 min. The mixtures were dropped on a mica surface and incubated for 5 to 10 min. The samples on the mica were scanned in Milli-Q water by HS-AFM (NanoExplorer, Research Institute of Biomolecule Metrology, RIBM). HS-AFM images were corrected for the background subtraction, followed by the automatic optimization of contrast and brightness by ImageJ software31.
Cell culture and treatments
Jurkat cells were cultured at 37°C in 5% CO2 in RPMI 1640 supplemented with 10% FBS (MP Biosceience). 293 cells were cultured at 37°C in 5% CO2 in DMEM/F-12 supplemented with antibiotics solution (A5955, Sigma-Aldrich) and 10% FBS. The CD61-expressing 293 cell was established as follows. 293 cells transfected with a pDIA-CD61 vector which carries an IRES-Azami Green fluorescnence protein, were provided by MBL. The cells were cultured under the same conditions as the 293 cells except for 500–1,000 μg/mL of G418 added to select the stable cell line. After three-time passages, single Azami Green positive cells were sorted into a 384 well culture plate by Fluorescence-Activated Cell Sorting, FACS Aria (BD bioscience). Clones with the high Azami Green fluorescence were then selected. The expression of the CD61 was confirmed by immunofluorescence staining with the anti-CD61 antibody (primary) and phycoerythrin-conjugated anti-mouse IgG (secondary) followed by FACS.
Avidity assays of antibody-containing Tri-RNPs on the 293 cells
The CD61-expressing 293 cells were plated into a 96 well plate with 4×104 cells per well. After 1 day incubation, the cells were incubated in 50 μL of the solution containing the anti-CD61 antibody or the control mouse IgG2a 6H3H11 provided by MBL at 37°C for 1 hour and washed with OptiMEM (life technologies) twice. 30 nM of L7Ae-Gβ1x2 and 10 nM of alexa 647-labeled Tri-RNAs were mixed and added to each well. After the 1 hour of incubation, the cells were washed with OptiMEM twice to remove remaining RNAs and L7Ae-Gβ1x2 and observed on RS-100 imager (Olympus), an automated cell imaging instrument, by a 627 nm LED and a 705/35 nm filter for excitation and emission, respectively.
Flowcytometry analysis of apoptosis by galectin-1-conjugated RNPs
The RNPs were prepared with the indicated protein and RNA as in the EMSA section except that OptiMEM was used instead of L7Ae buffer. For the experiment using lactose as a blocking agent for galectin-1 binding, lactose dissolved in OptiMEM was added to the RNA solutions at a final concentration of 30 mM. The annealed RNA was mixed with six times more amount of the L7Ae-Gal1 or L7Ae-Gal1β gently. 50 μL of the RNP solutions were transferred to 96 well plates and incubated at 37°C for more than 30 min. For the competition assays between Tri-RNP15 and 92, each RNP was separately prepared and mixed together in a 96 well plate. After the incubation, 50 μL of Jurkat cells washed by OptiMEM twice were added to the RNP solutions (final concentration of 1×106 cells/mL) and shaken for 10 sec on a microshaker (M-BR-024, TAITEC) gently. After 6 hours at 37°C, lactose (final concentration of 0.1 M) was added and the cell-clumps were carefully pippetted up and down to dissociate the galectins. The apoptotic and dead cells were stained by Pacific BlueTM Annexin V/SYTOX® AADvancedTM Apoptosis Kit (Invitrogen) according to the manufacturer's instructions. The stained cells were analyzed on FACS Aria (BD Bioscience). A 405 nm laser was used for excitation of the Pacific blue and 488 nm for the AADvanced dye. The 450/40 and 616/23 nm filter were used for collection of the emission. Collected data were analyzed by R32 with flowViz and flowCore packages33.
Quantification of the RNPs bound to the Jurkat cell surface
The RNP solutions were prepared with the indicated protein and RNA in the same way as the flowcytometry analysis section. Jurkat cells were stained in the culture medium supplemented with 10 μg/mL of Hoechst 33342 for 30 min followed by wash twice. The stained cells were re-suspended in OptiMEM supplemented with 20 μg/mL of Propidium Iodide, PI. 50 μL of the cell suspension were added into the 50 μL of RNP solutions in a 96 well plate and mixed for 10 sec on a microshaker. After incubation of 30 min at 37°C, the medium was replaced with fresh OptiMEM carefully. The nucleus and RNAs on Jurkat cells were visualized on RS-100 imager by using 365 nm LED and a 420 nm high band-pass filter for Hoechst 33342 and 530 nm LED and a 590 nm high band-pass filter for PI. The regions with the high fluorescent signal from Hoechst 33342 were defined as nuclei. The cells whose nucleus was stained with both Hoechst and PI were regarded as dead cells and excluded from the quantification. The fluorescent signals of PI in the areas 14 pixels within the nuclear envelope excluding the nuclei were regarded as the RNAs bound to a cell. The fluorescent signal was normalized by the number of the base pairs of the indicated Tri-RNA in order to obtain the relative number of RNA on a cell surface. To calculate the mean amount of RNA on cell surface, 100-3,000 cells were analyzed in each sample.
References
Bonnet, J., Yin, P., Ortiz, M. E., Subsoontorn, P. & Endy, D. Amplifying genetic logic gates. Science 340, 599–603 (2013).
Deniaud, D., Julienne, K. & Gouin, S. G. Insights in the rational design of synthetic multivalent glycoconjugates as lectin ligands. Org. Biomol. Chem. 9, 966–979 (2011).
Conway, A. et al. Multivalent ligands control stem cell behaviour in vitro and in vivo. Nat. Nanotechnol. 8, 831–838 (2013).
Bethani, I., Sk, Dikic, I. & Acker-Palmer, A. Spatial organization of transmembrane receptor signalling. EMBO J. 29, 2677–2688 (2010).
Holler, N. et al. Two adjacent trimeric Fas ligands are required for Fas signaling and formation of a death-inducing signaling complex. Mol. Cell. Biol. 23, 1428–1440 (2003).
Brewer, C. F., Miceli, M. C. & Baum, L. G. Clusters, bundles, arrays and lattices: novel mechanisms for lectin-saccharide-mediated cellular interactions. Curr. Opin. Struct. Biol. 12, 616–623 (2002).
Baer, K. et al. Dimerization-induced activation of soluble insulin/IGF-1 receptor kinases: an alternative mechanism of activation. Biochemistry 40, 14268–14278 (2001).
Shiroishi, M. et al. Efficient leukocyte Ig-like receptor signaling and crystal structure of disulfide-linked HLA-G dimer. J. Biol. Chem. 281, 10439–10447 (2006).
Ranzinger, J. et al. Nanoscale arrangement of apoptotic ligands reveals a demand for a minimal lateral distance for efficient death receptor activation. Nano Lett. 9, 4240–4245 (2009).
Koyfman, A. Y., Braun, G. B. & Reich, N. O. Cell-targeted self-assembled DNA nanostructures. J. Am. Chem. Soc. 131, 14237–14239 (2009).
Abdelmawla, S. et al. Pharmacological characterization of chemically synthesized monomeric phi29 pRNA nanoparticles for systemic delivery. Mol. Ther. 19, 1312–1322 (2011).
Kazane, S. A. et al. Self-assembled antibody multimers through peptide nucleic acid conjugation. J. Am. Chem. Soc. 135, 340–346 (2013).
Grabow, W. W. & Jaeger, L. RNA Self-Assembly and RNA Nanotechnology. Acc. Chem. Res. 47, 1871–1880 (2014).
Ohno, H. et al. Synthetic RNA-protein complex shaped like an equilateral triangle. Nat. Nanotechnol. 6, 116–120 (2011).
Derrick, J. P. & Wigley, D. B. The third IgG-binding domain from streptococcal protein G. An analysis by X-ray crystallography of the structure alone and in a complex with Fab. J. Mol. Biol. 243, 906–918 (1994).
Perillo, N. L., Pace, K. E., Seilhamer, J. J. & Baum, L. G. Apoptosis of T cells mediated by galectin-1. Nature 378, 736–739 (1995).
Pace, K. E., Lee, C., Stewart, P. L. & Baum, L. G. Restricted receptor segregation into membrane microdomains occurs on human T cells during apoptosis induced by galectin-1. J. Immunol. 163, 3801–3811 (1999).
Garner, O. B. & Baum, L. G. Galectin-glycan lattices regulate cell-surface glycoprotein organization and signalling. Biochem. Soc. Trans. 36, 1472–1477 (2008).
Belardi, B., O'Donoghue, G. P., Smith, A. W., Groves, J. T. & Bertozzi, C. R. Investigating cell surface galectin-mediated cross-linking on glycoengineered cells. J. Am. Chem. Soc. 134, 9549–9552 (2012).
Earl, L. A., Bi, S. & Baum, L. G. Galectin multimerization and lattice formation are regulated by linker region structure. Glycobiology 21, 6–12 (2011).
Bättig, P., Saudan, P., Gunde, T. & Bachmann, M. F. Enhanced apoptotic activity of a structurally optimized form of galectin-1. Mol. Immunol. 41, 9–18 (2004).
Cedeno-Laurent, F. et al. Development of a nascent galectin-1 chimeric molecule for studying the role of leukocyte galectin-1 ligands and immune disease modulation. J. Immunol. 185, 4659–4672 (2010).
Cho, M. & Cummings, R. D. Characterization of monomeric forms of galectin-1 generated by site-directed mutagenesis. Biochemistry 35, 13081–13088 (1996).
Miura, T. et al. Galectin-1beta, a natural monomeric form of galectin-1 lacking its six amino-terminal residues promotes axonal regeneration but not cell death. Cell Death Differ. 11, 1076–1083 (2004).
Rabinovich, G. A. et al. Molecular mechanisms implicated in galectin-1-induced apoptosis: activation of the AP-1 transcription factor and downregulation of Bcl-2. Cell Death Differ. 7, 747–753 (2000).
Kovács-Sólyom, F. et al. Mechanism of tumor cell-induced T-cell apoptosis mediated by galectin-1. Immunol. Lett. 127, 108–118 (2010).
Cironi, P., Swinburne, I. A. & Silver, P. A. Enhancement of cell type specificity by quantitative modulation of a chimeric ligand. J Biol Chem 283, 8469–8476 (2008).
Taylor, N. D., Way, J. C., Silver, P. A. & Cironi, P. Anti-glycophorin single-chain Fv fusion to low-affinity mutant erythropoietin improves red blood cell-lineage specificity. Protein Eng Des Sel 23, 251–260 (2010).
Lienert, F., Lohmueller, J. J., Garg, A. & Silver, P. A. Synthetic biology in mammalian cells: next generation research tools and therapeutics. Nat Rev Mol Cell Biol 15, 95–107 (2014).
Culler, S. J., Hoff, K. G. & Smolke, C. D. Reprogramming cellular behavior with RNA controllers responsive to endogenous proteins. Science 330, 1251–1255 (2010).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Ihaka, R. & Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comp. Graph. Stat. 5, 299–314 (1996).
Sarkar, D., Meur, N. L. & Gentleman, R. Using flowViz to visualize flow cytometry data. Bioinformatics 24, 878–879 (2008).
Acknowledgements
We are thankful for the collaboration of Dr. Hirohide Saito and Dr. Eriko Osada during the early stages of the research. We also thank MBL (Medical & Biological Laboratories Co., LTD) for providing the pDIA-CD61 plasmid and anti-CD61 antibodies and Dr. Eiry Kobatake (Tokyo Institute of Technology) for providing the Gβ1 plasmid. This work was supported by grants from JSPS (Number 23221011, 23116005) and JST (the International Cooperative Research Project [ICORP]).
Author information
Authors and Affiliations
Contributions
Y.F. and T.I. designed the project. Y.F., F.S. and T.I. evaluated the experimental results. Y.F. designed RNP structures and performed the whole experiments. H.O. designed RNP structures. R.F. and F.S. prepared materials and checked the reproducibility of the size-dependent apoptosis. Y.F., F.S. and T.I. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary Dataset 1
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Fujita, Y., Furushima, R., Ohno, H. et al. Cell-surface receptor control that depends on the size of a synthetic equilateral-triangular RNA-protein complex. Sci Rep 4, 6422 (2014). https://doi.org/10.1038/srep06422
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06422
This article is cited by
-
Protein-driven RNA nanostructured devices that function in vitro and control mammalian cell fate
Nature Communications (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
