
Abstract
Here we present the first report of a carbon-γ-Fe2O3 nanoparticle composite of mesoporous carbon, bearing COOH- and phenolic OH- functional groups on its surface, a remarkable and magnetically separable adsorbent, for the radioactive material emitted by the Fukushima Daiichi nuclear power plant accident. Contaminated water and soil at a level of 1,739 Bq kg−1 (134Cs and 137Cs at 509 Bq kg−1 and 1,230 Bq kg−1, respectively) and 114,000 Bq kg−1 (134Cs and 137Cs at 38,700 Bq kg−1 and 75,300 Bq kg−1, respectively) were decontaminated by 99% and 90% respectively with just one treatment carried out in Nihonmatsu city in Fukushima. Since this material is remarkably high performance, magnetically separable and a readily applicable technology, it would reduce the environmental impact of the Fukushima accident if it were used.
Similar content being viewed by others
Introduction
The magnitude 9.0 catastrophic earthquake and resulting tsunami on March 11, 2011, triggered the Fukushima Daiichi nuclear power plant accident, which is rated level seven. A very large amount of radioactive materials was vented into the atmosphere1 and subsequently caused serious radioactive contamination of the land over a wide region of Kanto and Tohoku, whereas western Japan was sheltered by mountain ranges2,3. In addition, a large quantity of water contaminated to a high level flowed out into the sea; the fisheries industry has suffered serious damage. The number of people who lived in the evacuation zones (areas with radiation levels higher than 50 mSv per year) was ca. 24,800 as of August 7th, 20134 and it is not anticipated that they will be able to return home for at least 6 years after the accident. The total area that needs radioactive decontamination is 26,700 ha (excluding forest). Total cesium-137(137Cs) depositions over the islands of Japan and the surrounding ocean (130–150°E and 30–46°N) were estimated to be more than 6.7 and 1.3 PBq, respectively2. Decontamination of soil and water containing radioactive materials is, therefore, a critical issue for people who lived in that region of Fukushima.
Since almost all of the cesium ions emitted by the plant are bonded and/or adsorbed to something (mainly clay) in the natural environment, an adsorbent must have several sizes of pore to sorb the clay particles and various surface functional groups such as OH- and COOH groups to bond ions associated with the cation-exchange mechanisms5. Therefore, ferric ferrocyanide (prussian blue)6, zeolite7,8, highly ordered mesoporous materials9 and carbon nanotubes10 are not suitable as a sorbent for decontamination under natural conditions, as reported from national decontamination pilot projects adopted by the Japan Atomic Energy Agency (Ministry of the Environment Government of Japan)11,12. Additionally, these materials and graphene oxide5 could not readily separate decontaminated soil from contaminated soil and water.
There are many reports of magnetic carbon sorbents, such as magnetic activated carbon13,14, magnetic carbon nanotubes15,16 and carbon-encapsulated magnetic nanoparticles17,18. Nanoparticles of various iron oxides, including magnetite (Fe3O4), maghemite (γ-Fe2O3) and hematite (α-Fe2O3)19,20,21, have attracted significant attention from both theoretical and applied scientists due to their broad range of applications and their unique physical properties as components of magnetic storage media22 and ferrofluids23. They have also been investigated as Lewis acid catalysts (in the Fe3+ state) in homogeneous catalysis24, as catalysts for carbon nanotube growth25, as contrast agents for magnetic resonance imaging26 and also for drug targeting27,28,29 and chemotherapy applications28,30. Nonetheless, there have been only a few reports concerning the synthesis of mesoporous carbon materials containing iron oxide nanoparticles31. The reported syntheses have followed two main routes32. One of these involves a one-pot, block-copolymer self-assembly strategy33,34,35. This depends on soft-template synthesis36 using the direct carbonization of a resol, a metal salt and block copolymers. The other route is based on a nanocasting technique37 which uses co-casting synthesis38. This synthesis employs a mesoporous template of silica or carbon, an impregnation technique, an appropriate carbon precursor and a metal source. Both of these synthetic routes are involve many distinct stages and researchers have had to work hard to prevent the leaching and oxidation of deposited metal nanoparticles during the complicated procedures. The complexity of the procedure and the expense of the carbon source has hindered the production of a commercially viable magnetically separable mesoporous carbon material.
Herein, a new, simple and robust method for the preparation of a mesoporous carbon-γ-Fe2O3 nanoparticle composite (MCNC) is presented and this material's significant ability to remove radioactive material from contaminated soil and water are reported. This material can be directly synthesized in a one-pot, non-templating and self-sustaining reaction using carboxymethyl cellulose (CMC) as an inexpensive carbon source. The adsorption mechanism and performance of MCNC is discussed based on characterization, a laboratory scale experiment using a standardised solution of radioisotope cesium (Cs) and strontium (Sr) and a demonstration experiment in Nihonmatsu city in Fukushima, Japan.
Results
Synthesis
CMC is an anionic, semisynthetic water-soluble polymer and was used as an inexpensive carbon source for the synthesis of the catalyst. CMC is a cellulose derivative in which a quantity of the hydroxyl groups on the glucose repeating units have been replaced with carboxymethyl groups. In the synthetic process, CMC (10 g) was mixed with an iron(III) nitrate enneahydrate solution (1 L) prepared within the concentration range of 1.25–15.0 g L−1. Upon mixing, a red gel appeared in the solution, which was subsequently dried at 60°C for 5 days. During this step, nitric acid is produced by decomposition of iron(III) nitrate enneahydrate solution when the solution is heated above approximately 50°C39. When producing the MCNC using a heating mantle under N2 flow, a pre-carbonization or intra-condensation process in the CMC polymer is initiated in the temperature range of 150–220°C and is subsequently propagated as a self-sustaining reaction by oxidation with residual nitric acid (producing an internal temperature of 100–125°C) in which Fe(NO3)3·9H2O is decomposed to γ-Fe2O3. The final yields of the product are around 20% of the amount of raw material excluding added water for all concentrations of iron(III) nitrate enneahydrate solution (Figure S1). The yields of the precursors of MCNC (relative to the amount of raw material excluding added water) have a linear negative relationship to the concentration of iron(III) nitrate enneahydrate solution (Figure S1).
Structure
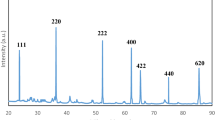
Figures 1a and b provide the powder X-ray diffraction (XRD) patterns and Raman spectra of the prepared material. The XRD pattern of MCNC exhibits a broad C (002) diffraction peak at 2θ angles of 10°–30°, attributed to carbon with a very low degree of graphitization40. Diffraction peaks within the range 30°–75° can be assigned to either γ-Fe2O3 or Fe3O4 and both these materials exhibit very weak, broad diffraction peaks characteristic of iron oxide, indicating the presence of nanoscale iron oxide particles38. This result can be attributed to the partial reduction in iron oxide nanoparticles owing to the nanoscale carbothermic reaction that occurs at the interface of the iron oxide nanoparticles and nanoscale carbon41,42. Sodium carbonate is also observed as a by-product of the synthesis, although this contaminant is completely removed by rinsing with hot distilled water (Figure 1a). In the Raman spectrum (Figure 1b), the intensity ratio of the D band (1,350 cm−1, A1g D breathing mode) to the G band (1,580 cm−1, E2g G mode) is 0.80, indicating that the average graphene dimension in the carbon is approximately 1 nm43,44.

Structures of MCNC (concentration of iron(III) nitrate enneahydrate solution: 5.0 g L−1).
(a) The XRD pattern of the samples. The diffraction peaks can be assigned to γ-Fe2O3 (JCPDS card No. 39-1346) or Fe3O4 (JCPDS card No. 19-0629). (b) Raman spectrum of the sample and (c) Mössbauer spectra at 293 K and 78 K.
The Mössbauer spectra at 293 K and 78 K are shown in Figure 1c. The room temperature spectrum exhibits what appears to be a broad singlet peak as the main feature. The overall spectrum can be described as being composed of a doublet and a broad sextet, which indicates a wide range of iron oxide particle sizes45. The Mössbauer effect parameters are very different from those expected based on the results of XRD analysis of γ-Fe2O3 or Fe3O4 content (see Table S1 for a summary of Mössbauer data), since the Mössbauer data indicate that the majority of the nanoparticles are paramagnetic. The results suggest that this material generates paramagnetic relaxation spectra due to the presence of superparamagnetic particles in the respective matrices46.
Since the relaxation time depends on measurement temperature, a Mössbauer spectrum was also acquired at liquid nitrogen temperatures (78 K) with the aim of observing a sextet peak. The spectrum at this temperature does indeed exhibit two sextets and a doublet. The strong doublet in this spectrum provides evidence for the presence of a relatively large amount of superparamagnetic material and/or paramagnetic Fe3+. The four outer peaks of the sextet have tails that extend inwards, suggesting an additional component to the spectrum. This may occur either because the internal magnetic spectrum appears smaller due to the effects of relaxation, or because of the internal magnetic field distribution, although these two causes essentially involve the same phenomena. Judging from the Mössbauer effect parameters (Table S1), MCNC is more likely to contain γ-Fe2O3, although the parameters of Fe3+ are very similar for both γ-Fe2O3 and Fe3O4 at room temperature. At low temperature, however, the spectrum of γ-Fe2O3 is almost unchanged, while that of Fe3O4 varies significantly between room temperature and 78 K due to a Verwey transition at approximately 120 K. At 78 K, the magnetic field distribution analysis was carried out under the assumption that the tails of the peaks were entirely attributable to magnetic field distribution. The mode value of the internal magnetic field distribution was found to be 502 kOe and there were no significant local maxima (Figure S2). Moreover, the peak located near the center of the spectrum, which is considered to indicate a paramagnetic component, could be interpreted as indicating a small magnetic field resulting from a single component. Consequently, the spectrum of MCNC can be interpreted as almost entirely due to a single factor, that being the presence of γ-Fe2O3.
The valences of iron ions may readily be established by X-ray photoelectron spectroscopy (XPS) analysis and thus this technique serves as a means of distinguishing γ-Fe2O3 from Fe3O4. Magnetite contains Fe2+ and Fe3+, both of which contribute to the Fe 2p3/2 spectrum as two overlapping components38. Typically, the primary Fe 2p3/2 for Fe3O4 appears at 710.4 eV47, slightly below the peak associated with Fe2O3 at 710.8 eV, and, based on Figure 2a, it is obvious that there is no Fe3O4 phase in either material. In addition, a split is observed in the main peak, which clearly differentiates γ-Fe2O3 from α-Fe2O338. These spectra also offer no evidence for the presence of a bond between Fe and C.

Surface analyses of MCNC.
(a) Fe 2p3/2 and (b) O 1s XPS spectra (concentration of iron(III) nitrate enneahydrate solution: 5.0 g L−1). (c) 13C-DD/MAS NMR spectrum (* denotes spinning side bands, concentration of iron(III) nitrate enneahydrate solution: 0.5 g L−1).
As seen in the O 1s spectra (Figure 2b), the peaks are somewhat asymmetric because of the presence of oxygen-containing impurities in the carbon framework. The reported values for the O 1s binding energies of α-Fe2O3 and Fe3O4 are identical at approximately 530.0 eV, slightly below that of γ-Fe2O3 (530.6 eV)47. The value of 531.7 eV obtained in the O 1s spectra is a good match with the expected value for γ-Fe2O3 and the lack of a shoulder peak on the lower energy side establishes the absence of Fe3O4 and α-Fe2O3.
The 13C dipolar decoupling magic angle spinning nuclear magnetic resonance (13C DD/MAS NMR) spectrum is shown in Figure 2c. Since the signal was greatly affected by iron oxide, the concentration of the iron(III) nitrate enneahydrate solution for the sample for this measurement was fixed at 0.5 g L−1. This broad spectrum contains a number of readily identifiable peaks at 132 (polycyclic aromatic carbons), 156 (phenolic OH) and 195 (COOH) ppm43,48,49. This result corresponds well to C 1s spectra from XPS analysis, which shows peaks attributed to C-C(H), C-O and C = O (284.5, 285.7 and 287.5 eV, respectively, Figure S3). The obtained material contains a low concentration of sp3 carbon atoms (50 ppm).
Magnetic properties
The magnetic properties of the obtained material at room temperature, measured using a vibration sample magnetometer (VSM) are shown in Figure 3a. The Ms values of MCNC are seen to increase with increasing concentrations of the iron nitrate solution. These curves are nearly superimposable as the field is cycled between −17 and 17 kOe (Table S2) indicating superparamagnetic behavior19. However, since the coercive force is too large for typical superparamagnetic materials, it seems that the obtained materials cannot be fully superparamagnetic. In measurements using a superconducting quantum interference device (SQUID), the blocking temperature of the sample was determined to be 22.5 K (Figure S4)50. Since the iron oxide nanoparticles have been dispersed in a carbon matrix, inter-particle interactions were impaired, leading to a broking temperature that was lower than those previously measured for γ-Fe2O3 nanoparticle46,50.

Magnetic properties and morphologies of MCNC.
(a) Hysteresis curves at room temperature (concentration of iron(III) nitrate enneahydrate solution: black 1.25 g L−1; green, 2.50 g L−1; blue, 5.00 g L−1; pink, 10.0 g L−1; red, 15.0 g L−1). (b) SEM image and (c) TEM image (concentration of iron(III) nitrate enneahydrate solution: 5.0 g L−1).
Morphology
The particle size ranges (Figures 3b) and Brauner-Emmett-Teller (BET) surface areas are estimated to be 2–100 μm and 36–152 m2 g−1 (Table S3), respectively. The particle sizes can also be readily modified simply by pulverization of the materials. The data in Table S3 demonstrate that the BET surface area generally increases with increasing concentration of the iron(III) nitrate enneahydrate solution used in the synthesis. This result also can be attributed to the nanoscale carbothermic reaction described previously. The N2 adsorption-desorption isotherm features a narrow hysteresis loop with parallel adsorption and desorption branches (Figure S5a), suggesting that mesopores and micropores coexist51. The total pore volume and the average size of ideal cylindrical pores, based on the BJH (Barrett–Joyner–Halenda) method52, are in the ranges of 0.10–0.16 cm3 g−1 and 4–12 nm, respectively (Table S3, Figure S5b). Although the average size of the ideal cylindrical pores is reduced when the concentration of iron(III) nitrate enneahydrate solution is increased, the total pore volumes increased with increasing solution concentration. This porous structure may be related to the aforementioned nanoscale carbothermic reaction.
Figures 3c presents a transmission electronic microscopy (TEM) image. Analysis of the sample via field emission TEM energy dispersive X-ray spectrometry (FE-TEM/EDS) revealed that this material includes distributed iron oxide (5–10 nm in size) within its carbon matrix and also demonstrated the presence of Fe, O, Na and C. In general, iron oxide nanoparticles smaller than approximately 20 nm display superparamagnetic behavior at room temperature (the critical size for maghemite is 10 nm)19. Therefore, the iron oxide particles in the material synthesized in this work should exhibit superparamagnetic behavior.
Chemical adsorption site
Figure S6 shows the ammonia temperature programmed desorption (NH3-TPD) profiles of MCNC. The samples show a broad desorption signal in the region of 100–350°C, indicating a wide distribution of surface functional groups due to the presence of graphene sheets bearing the phenolic OH and COOH groups previously identified from the NMR and XPS (C 1s) spectra of MCNC. The peak temperatures decrease with increasing concentrations of iron nitrate solution, with a maximum of 196°C at 1.25 g L−1. The total amount of NH3 desorbed from the MCNC, a measure of the total number of chemical adsorption sites, increases with increasing concentrations of iron(III) nitrate solution, reaching a maximum at a concentration of 82.8 μmol g−1, whereas further increases in the concentration of the iron nitrate solution tend to decrease the total desorption (Table S4). The higher specific surface area of the material synthesized using higher iron nitrate concentrations resulted in a high number of acid sites (Table S4). Since materials with more available carbon are able to bind a greater quantity of functional groups, the acid strength (Table S5) was great.
Adsorbent performance
The concentration dependence of Cs (pH = 5.7) and Sr (pH = 3.56–4.74) adsorption with MCNC is shown in Figure 4a. The adsorbent demonstrated superior performance on radioisotopes of less than 0.05 mM concentration, 97% and 99% respectively. The capacity of MCNC for Cs adsorption can be estimated as 0.488 mmol g−1. On the other hand, the number of chemical adsorption sites is estimated to be 0.0455 mmol g−1 from the results of NH3-TPD. Consequently, these results show that most of the adsorption is physical adsorption in this reaction. The kinetics of Cs removal at pH = 5.7 with MCNC are shown in Figure S7, indicating that a steady state condition was achieved within 1 min. This fast kinetics is favorable for practical applications of MCNC for removal of cationic impurities. Figure 4b shows the pH dependence of the adsorption of Cs and Sr by MCNC. Both of the radioisotopes demonstrate typical S-shaped pH-edges for cations. A high rate of adsorption of Cs is achieved at pH > 2.5. This result suggests that MCNC would be useful in contaminated natural waters with a broad range of pH values. A high rate of adsorption of Sr occurred at pH > 6. These adsorption phenomena can be attributed to the substituent effect on aromatic carbon. Since two carboxyl groups need to bond to Sr, adsorption of Sr shows sensitivity to proton concentration.

Adsorption performances of MCNC (concentration of iron(III) nitrate enneahydrate solution: 5.0 g L−1).
(a) Dependence on concentration. Carbon, 1 g; time, 1 h; agitating speed, 150 rpm; CsCl solution, 10 mL; particle size, under 150 μm. SrCO3 in 0.1 mol L−1 HNO3 solution, 10 mL; particle size, under 150 μm; wavelength, 407.7 nm in ICP analysis. (b) Dependence of pH. Carbon, 2 g; time, 1 h; agitating speed, 150 rpm; CsCl solution concentration, 0.0075 mM (20 mL); particle size, under 150 μm. Buffer solution: hydrochloric acid (triangle), acetic acid/sodium acetate (diamond), Britton-Robinson (square). (c) Demonstration experiments with contaminated water (carbon, 5.0 g; contaminated water, 1 L) and (d) contaminated soil (carbon, 25.0 g; contaminated soil, 100 g; distilled water, 500 mL; agitation, 0.5 h).
Figure S8 show the iodine and methylene blue adsorption performances of MCNC tested in relation to JIS K 1474. The adsorption ratio of iodine by MCNC was 230 mg g−1 when the residual concentration of iodine was 2.5 g L−1 (Figure S8a). This result reveals that relatively small atoms also can be adsorbed by MCNC. The adsorbed ratios of methylene blue were 9.7 (particle size: 100–150 μm) and 12.7 mg g−1 (particle size: under 100 μm) when the residual concentration of methylene blue was 0.24 mg L−1 (Figure S8b and c, extrapolating: unmeasurable level), which revealed that MCNC can adsorb large basic molecules and that adsorption of methylene blue depends on the particle size of the adsorbent.
Figure S9 shows the results of XPS analysis in relation to cesium, strontium, iodine and methylene blue adsorption. As seen in the Cs 3d and Sr 3d spectra (Figure S9a and b), peaks are observed at 726 and 738 eV and 135 eV. These results demonstrate that Cs and Sr bond with carboxyl groups on the surface of the material. In the case of iodine (I 3d), peaks are also observed around 630 eV (Figure S9c). On the other hand, peaks are not observed in relation to methylene blue (Cl 2p, S 2p) (Figure S9d). This suggests that the basic molecule is adsorbed into the pore by physical adsorption.
Demonstration experiment
Figure 4c and d show the results of a verification test on contaminated water obtained from a fire-prevention water tank and on soil collected from under the drainpipe outlet of a house in Nihonmatsu city, respectively. Contaminated water containing suspended substance (SS) at a level of 1,739 Bq kg−1 (134Cs and 137Cs of 509 Bq kg−1 and 1,230 Bq kg−1, respectively) was decontaminated to 14 Bq kg−1 (99% decontamination) with just one treatment (Figure 4c). All of the Cs is bonded to carboxyl groups of the SS, which is made up of decaying vegetation. The sorbent formed ‘floc’ with SS and consequently the generated floc was easily separated with a magnet.
Contaminated soil collected from under the drainpipe outlet of the house was contaminated at a level of 114,000 Bq kg−1 (134Cs and 137Cs of 38,700 Bq kg−1 and 75,300 Bq kg−1, respectively) and was decontaminated to 3,980 and 7,520 Bq kg−1 (90% decontamination) respectively with just one treatment using a solid-solid phase separation technique (Figure 4d). In the case of soil, all of the Cs is bonded with clay. The same results have been obtained in an experiment with non-magnetic soil (the percentage of magnetic soil in the verification test was 30%) and so our results do not indicate that the material can only separate magnetic soils. Figure S10 shows the SEM images of the sorbent and soil that were used in the verification test and of MCNC bonded to clay particles. The size of the clay particles was several μm to 50 μm. An EDX analysis revealed that the soil contained Fe, Al, Si, K and Ba. Contaminated clay particles cover the surface of the MCNC and as a result, a decontamination effect is seen.
Discussion
Ferric ferrocyanide6, zeolite7,8 highly ordered mesoporous materials9 and carbon nanotubes10 are important sorbents for ionic state radioactive material, but such materials do not function properly for actual decontamination as reported from national decontamination pilot projects11,12. Additionally, these materials and graphene oxide5 could not readily separate decontaminated soil from contaminated soil and water because they do not have magnetic properties. Most conventional decontamination methods for contaminated soil require the treatment of contaminated water including SS and flocculant and/or super filters have a central role in these systems11,12. Nevertheless, the decontamination rate of these conventional methods remains in the region of 80%. In our new method, contaminated water and soil at a level of 1,739 Bq kg−1 and 114,000 Bq kg−1 were decontaminated by 99% and 90% respectively with just one treatment, as shown in Figure 4c and d. Since Cs bonds to clay or aquatic humic substances in ponds and lakes4,11,12, varied surface functional groups and pores of varied sizes are absolutely imperative in an adsorbent intended for use to decontaminate soil and water (containing SS) and ionic state radioactive Cs. On this point, our new material has the advantages that the adsorbent has pores of varied sizes (Figure S5 and Table S3) and with varied functional groups (Figure 2 and S6, Table S4) and also has magnetic properties (Figure 3a and Table S2).
The decontamination of fields and of the sludge on the bottom of ponds, lakes and rivers is very important for the safety of the water supply and of food1,2,3,4,11,12. These issues impact many aspects of life, such as agriculture, especially wet-field rice agriculture, fishing and municipal drinking water supplies. It is clear that our material is extremely useful not only for the decontamination of water and soil but also to ensure peace of mind and the safety of the food and water supply. The adsorbent could support reactor decommissioning, because a robot could spread it in areas with high radiation levels and collect it with an electromagnet. The necessary equipment would be compact and portable. It could potentially also be used to remove uranium contamination at abandoned mine sites53 or to decontaminate the seabed.
Methods
Characterization
Structural information for the material was obtained by XRD (CuKα, λ = 1.54056 Å, 40 kV, 200 mA (focusing method), Rigaku RINT1500), Raman spectroscopy (Jobin Yvon T-64000), 57Fe Mössbauer spectroscopy (Toray Research Center), XPS (AlKα, 1486.6 eV, ULVAC-PHI Quantera SXM) and 13C DD/MAS NMR (JEOL JNM-ECX400) at room temperature. The magic angle was controlled at a 15 kHz sample tube (4 mm) rotation rate and the background signal was acquired using KBr under the same conditions. Mössbauer spectra were recorded on a Wissel spectrometer in the constant acceleration mode (transmission method), using a 57Co(Rh) source (1.85 GBq). Samples for Mössbauer analysis were prepared by mixing 80 mg of material and 10 mg of high-purity polyethylene, followed by pressing at 14.7 MPa. The elemental compositions of the samples were determined using two methods. Precise values for C, H and N were obtained using an elemental analyzer (J-Science Lab. JM-10), while values for O, Na and Fe were estimated from XPS analysis. The morphology of the synthesized material was determined by scanning electron microscopy (SEM, Hitachi High-Technologies S-3000N), nitrogen adsorption and desorption isotherms (including BET surface area measurements, Quantachrome Instruments NOVA 4200e) and FE-TEM/EDS (Topcon Technohouse EM002BF). Magnetization measurements of the material were performed using VSM (Toei Industry VSM-15) at room temperature (25°C) and SQUID (Quantum Design SQUID magnetometer MPMS3) at 5 K to 300 K (1000 Oe). The adsorption site concentration of MCNC was evaluated by the NH3-TPD method using a quadrupole mass analyzer (m/z = 16, BEL Japan TPD-1-ATw). Samples (ca. 0.05 g) were preheated at 400°C for 1 h (using a ramp rate of 10°C min−1) under helium flow (50 cm3 min−1) to remove residual water and other volatile species. The samples were then cooled to 100°C and held at that temperature while exposed to ammonia. An NH3/He gas mixture (0.5% ammonia) was passed over the sample for 0.5 h at a flow rate of 100 cm3 min−1 and the system was subsequently purged with pure helium at 50 cm3 min−1 for 0.5 h to remove physically adsorbed ammonia. Measurements of ammonia desorption were then performed using a ramp rate of 10°C min−1.
Adsorbent performance
In the laboratory experiments, we used CsCl and SrCO3 in 0.1 mol L−1 HNO3 solution (Wako) as radioisotope cesium and strontium, respectively. The samples were evaluated with inductively-coupled plasma (ICP)-mass analysis (Hitachi high-tech science SPQ9000) for Cs and Sr (pH dependence) and ICP analysis (Thermo Fisher Scientific iCAP6000) for Sr (concentration dependence, wavelength: 407.7 nm). Initial concentrations for tests of dependence on concentration were 0.7509, 0.0751, 0.0075 and 0.0015 mM for Cs (pH = 5.7) and 0.0457, 0.0228, 0.0114, 0.0057 and 0.0029 mM for Sr (pH = 3.56–4.74). The initial total concentration of radioisotopes in the kinetics experiments and pH-dependence tests was 0.0075 mM for Cs and 0.0114 mM for Sr. Hydrochloric acid buffer solution (Cs: pH = 1.10, 2.07, 3.05, Sr: pH = 2.94), sodium acetate buffer solution (Cs: pH = 3.11, 4.41, 5.73, Sr: pH = 3.05, 4.07, 5.71) and Britton-Robinson buffer solution (Cs: pH = 2.23, 3.28, 4.38, 5.86, 7.09, 8.19, 10.13, 11.65, Sr: pH = 3.12, 4.22, 6.20, 8.10, 10.13, 12.00) were used to attenuate the CsCl and SrCO3 solutions a thousandfold at several pH. 2 g of sorbent (concentration of iron nitride, 5 g L−1; particle size, under 150 μm) was added to 20 mL of solution and then stirred for 60 minutes before ICP analysis.
Iodine and methylene blue adsorption performance were examined in relation to JIS K 1474. The amounts of adsorbent were as follows: 0.5, 1.0, 2.0, 3.0 g for iodine (particle size, under 150 μm) and 2.0, 2.4, 2.5, 2.6 g (particle size: 100–150 μm) and 1.8, 1.9, 2.0 (particle size: under 100 μm) for methylene blue. A spectral photometer (wavelength: 665 nm, Shimazu UV-3100PC) was used in the methylene blue experiment.
Radiological dose was evaluated with a germanium semiconductor detector (Canberra GC 4020 7500SL, measurement time: 2,000 seconds, detection limit: 8–6 Bq kg−1) using a U-8 holder and scintillation survey meter (Hitachi Aloka Medical TCS-172B) in the demonstration experiment with contaminated water and soil in Nihonmatsu city (latitude and longitude of the collection sites are 37 degrees 38′ 22″ N and 140 degrees 26′ 09″ E and 37 degrees 38′ 36″ N and 140 degrees 26′ 02″ E, respectively). The concentration of SS in contaminated water obtained from the fire-prevention water tank was 2.8 g L−1 (the levels of 134Cs and 137Cs are 91,500 Bq kg−1 and 191,000 Bq kg−1 respectively when the SS is in a dry condition). 5.0 g of sorbent was mixed in 1 L of contaminated water and then left to stand for one a day. Decontaminated water and the generated floc were separated with a magnet.
Contaminated soil was collected from under the drainpipe outlet of a house (moisture content was 57.7% of the dry base). Soil size distribution based on JIS A 1202–1205, 1210, 1216–1218, 1223–1228, was as follows: gravel, 9.1%; sand, 31.1%; silt, 39.9%; clay, 20.0%. 25 g of the sorbent was mixed with 100 g of contaminated soil and 500 mL of water and then stirred for 30 minutes. The sorbent that we were able to separate with a magnet was separated from the mixture before drying at 105°C for 24 hours. The obtained soil-sorbent cake was crushed to enable the separation of decontaminated soil and sorbent with a magnet. The decontaminated soil was rinsed with 200 mL of water to enable the separation of the adsorbent with a magnet. After drying at 105°C for 24 hours, the decontaminated soil was analyzed.
References
Chino, M. et al. Preliminary Estimation of Release Amounts of 131I and 137Cs Accidentally Discharged from the Fukushima Daiichi Nuclear Power Plant into the Atmosphere. J. Nucl. Sci. Technol. 48, 1129–1134 (2011).
Yasunari, T. J. et al. Cesium-137 Deposition and Contamination of Japanese Soils due to the Fukushima Nuclear Accident. Proc. Nat. Acad. Sci. USA. 108, 19530–19534 (2011).
Butler, D. Radioactivity Spreads in Japan. Nature 471, 555–556 (2011).
Cabinet Office of Japan, Areas to which Evacuation Orders have been Issued (2013). (first issued August 7 2103, updated on August 13, 2013) Date of access: 03/02/2014. Available at: “http://www.meti.go.jp/english/earthquake/nuclear/roadmap/pdf/20130807_01.pdf” and “http://www.meti.go.jp/earthquake/nuclear/pdf/131009/131009_02a.pdf” (in Japanese) (updated on October 11 2013).
Romanchuk, A. Y., Slesarev, A. S., Kalmykov, S. N., Kosynkini, D. V. & Tour, J. M. Graphene Oxide for Effective Radionuclide Removal. Phys. Chem. Chem. Phys. 15, 2321–2327 (2013).
Kouřím, V., Rais, J. & Million, B. Exchange Properties of Complex Cyanides—I: Ion Exchange of Caesium of Ferrocyanides. J. Inorg. Nucl. Chem. 26, 1111–1115 (1964).
Ames, L. L., Jr The Cation Sieve Properties of Chlinoptilolite. Am. Min. 45, 689–700 (1960).
Ames, L. L., Jr Cation Sieve Properties of the Open Zeolite, Chabazite, Mordenite, Erionite and Chlinoptilolite. Am. Min. 46, 1120–1131 (1961).
El-Safty, S. A. Development of HOM Nanomaterials for Detection Simple Removal of Radioactive Cesium137 in Water in the Optical Ion-Sensitive Removal of Mercury Ions. Sci. Acad. of Tsukuba 24, 10–11 (2013).
Tsuruoka, S. et al. Intensive Synergetic Cs Adsorbent Incorporated with Polymer Spongiform for Scalable Purification without Post Filtration. Mater. Express 3, 21–29 (2013).
Shiratori, Y. & Tagawa, A. (Japan Atomic Energy Agency), Report of the Results of the Decontamination Technology Demonstration Test Project. FY 2011 “Decontamination Technology Demonstration Test Project”., (2012) Date of access: 03/02/2014. Available at: “http://www.jaea.go.jp/fukushima/decon04/english/3-1%20Decontamination_Technology_Demonstration_Test_Project.pdf” and “http://fukushima.jaea.go.jp/pdf/techdemo_report.pdf” (in Japanese) (updated on October 23 2012).
Japan Atomic Energy Agency, Report of the Results of the Decontamination Technology Demonstration Test Projects., (2013) Date of access: 03/02/2014. Available at: “http://fukushima.jaea.go.jp/initiatives/cat01/pdf07/130627.pdf” (in Japanese).
Oliveiraa, L. C. A. et al. Activated Carbon/Iron Oxide Magnetic Composites for the Adsorption of Contaminants in Water. Carbon 40, 2177–2183 (2002).
Wang, C., Liu, Q., Cheng, X. & Shen, Z. Adsorption and Desorption of Gold on the Magnetic Activated Carbon. J. Mater. Sci. Technol. 10, 151–153 (1994).
Gong, J. L. et al. Removal of Cationic Dyes from Aqueous Solution Using Magnetic Multi-Wall Carbon Nanotube Nanocomposite as Adsorbent. J. Hazard. Mater. 164, 1517–1522 (2009).
Pardasani, D., Kanaujia, P. K., Purohit, A. K., Shrivastava, A. R. & Dubey, D. K. Magnetic Multi-Walled Carbon Nanotubes Assisted Dispersive Solid Phase Extraction of Nerve Agents and Their Markers from Muddy Water. Talanta 86, 248–255 (2011).
Herrmann, I. K. et al. Blood Purification Using Functionalized Core/Shell Nanomagnets. Small 6, 1388–1392 (2010).
Bystrzejewski, M., Pyrzyńska, K., Huczko, A. & Lange, H. Carbon-Encapsulated Magnetic Nanoparticles as Separable and Mobile Sorbents of Heavy Metal Ions from Aqueous solutions. Carbon 47, 1201–1204 (2009).
Teja, A. S. & Koh, P. Y. Synthesis, Properties and Applications of Magnetic Iron Oxide Nanoparticles. Prog. Cryst. Growth Charact. Mater. 55, 22–45 (2009).
Wang, L. L. & Jiang, J. S. Preparation of Fe3O4 Spherical Nanoporous Particles Facilitated by Polyethylene Glycol 4000. Nanoscale Res. Lett. 4, 1439–1446 (2009).
Asuha, S., Zhao, S., Jin, X. H., Hai, M. M. & Bao, H. P. Effects of Synthetic Routes of Fe–Urea Complex on the Synthesis of γ-Fe2O3 Nanopowder. Appl. Surf. Sci. 255, 8897–8901 (2009).
Dhara, S., Rastogi, A. C. & Das, B. K. Direct Deposition of Highly Coercive Gamma Iron Oxide Thin Films for Magnetic Recording. J. Appl. Phys. 74, 7019–7021 (1993).
Raj, K. & Moskowitz, R. Commercial Applications of Ferrofluids. J. Magn. Magn. Mater. 85, 233–245 (1990).
Shi, F. et al. Tuning Catalytic Activity between Homogeneous and Heterogeneous Catalysis: Improved Activity and Selectivity of Free Nano-Fe2O3 in Selective Oxidations. Angew. Chem. Int. Ed. 46, 8866–8868 (2007).
Fleaca, C. T. et al. Magnetic Properties of Core-Shell Catalyst Nanoparticles for Carbon Nanotube Growth. Appl. Surf. Sci. 255, 5386–5390 (2009).
Pouponneau, P., Leroux, J. C. & Martel, S. Magnetic Nanoparticles Encapsulated into Biodegradable Microparticles Steered with an Upgraded Magnetic Resonance Imaging System for Tumor Chemoembolization. Biomaterials 30, 6327–6332 (2009).
Oh, W. K., Yoon, H. & Jang, J. Size Control of Magnetic Carbon Nanoparticles for Drug Delivery. Biomaterials 31, 1342–1348 (2010).
Rudge, S. R., Kurtz, T. L., Vessely, C. R., Catterall, L. G. & Williamson, D. L. Preparation, Characterization and Performance of Magnetic Iron-Carbon Composite Microparticles for Chemotherapy. Biomaterials 21, 1411–1420 (2000).
Bergemann, C., Müller-Schulte, D., Oster, J., à Brassard, L. & Lübbe, A. S. Magnetic Ion-Exchange Nano- and Microparticles for Medical, Biochemical and Molecular Biological Applications. J. Magn. Magn. Mater. 194, 45–52 (1999).
Jordan, A. et al. Presentation of a New Magnetic Field Therapy System for the Treatment of Human Solid Tumors with Magnetic Fluid Hyperthermia. J. Magn. Magn. Mater. 225, 118–126 (2001).
Sevilla, M., Valle-Vigón, P., Tartaj, P. & Fuertes, A. B. Magnetically Separable Bimodal Mesoporous Carbons with a Large Capacity for the Immobilization of Biomolecules. Carbon 47, 2519–2527 (2009).
Wang, Z., Liu, X., Lv, M. & Meng, J. Simple Synthesis of Magnetic Mesoporous FeNi/Carbon Composites with a Large Capacity for the Immobilization of Biomolecules. Carbon 48, 3182–3189 (2010).
Wang, D. W., Li, F., Lu, G. Q. & Cheng, H. M. Synthesis and Dye Separation Performance of Ferromagnetic Hierarchical Porous Carbon. Carbon 46, 1593–1599 (2008).
Huang, C. H., Doong, R. A., Gu, D. & Zhao, D. Dual-Template Synthesis of Magnetically-Separable Hierarchically-Ordered Porous Carbons by Catalytic Graphitization. Carbon 49, 3055–3064 (2011).
Yao, J., Li, L., Song, H., Liu, C. & Chen, X. Synthesis of Magnetically Separable Ordered Mesoporous Carbons from F127/[Ni(H2O)6](NO3)2/Resorcinol-Formaldehyde Composites. Carbon 47, 436–444 (2009).
Zhai, Y. et al. Soft-Template Synthesis of Ordered Mesoporous Carbon/Nanoparticle Nickel Composites with a High Surface Area. Carbon 49, 545–555 (2011).
Lu, A. H. et al. Nanoengineering of a Magnetically Separable Hydrogenation Catalyst. Angew. Chem. Int. Ed. 43, 4303–4306 (2004).
Dong, X., Chen, H., Zhao, W., Li, X. & Shi, J. Synthesis and Magnetic Properties of Mesostructured γ-Fe2O3/Carbon Composites by a Co-Casting Method. Chem. Mater. 19, 3484–3490 (2007).
Guo, Z. et al. Effects of Iron Oxide Nanoparticles on Polyvinyl Alcohol: Interfacial Layer and Bulk Nanocomposites Thin Film. J. Nanopart. Res. 12, 2415–2426 (2010).
Tsubouchi, N., Xu, C. & Ohtsuka, Y. Carbon Crystallization during High-Temperature Pyrolysis of Coals and the Enhancement by Calcium. Energ. Fuel. 17, 1119–1125 (2003).
Cao, F., Chen, C., Wang, Q. & Chen, Q. Synthesis of Carbon–Fe3O4 Coaxial Nanofibres by Pyrolysis of Ferrocene in Supercritical Carbon Dioxide. Carbon 45, 727–731 (2007).
Chen, W., Pan, X., Willinger, M. G., Su, D. S., & Bao, X. Facile Autoreduction of Iron Oxide/Carbon Nanotube Encapsulates. J. Am. Chem. Soc. 128, 3136–3137 (2006).
Suganuma, S. et al. Hydrolysis of Cellulose by Amorphous Carbon Bearing SO3H, COOH and OH Groups. J. Am. Chem. Soc. 130, 12787–12793 (2008).
Ferrari, A. C. & Robertson, J. Interpretation of Raman Spectra of Disordered and Amorphous Carbon. Phys. Rev. B 61, 14095–14107 (2000).
Ni, Y., Ge, X., Zhang, Z. & Ye, Q. Fabrication and Characterization of the Plate-Shaped γ-Fe2O3 Nanocrystals. Chem. Mater. 14, 1048–1052 (2002).
Tronc, E. et al. Magnetic Behaviour of γ-Fe2O3 Nanoparticles by Mössbauer Spectroscopy and Magnetic Measurements. Hyperfine Interact. 95, 129–148 (1995).
Fujii, T. et al. In situ XPS Analysis of Various Iron Oxide Films Grown by NO2-Assisted Molecular-Beam Epitaxy. Phys. Rev. B 59, 3195–3202 (1999).
Toda, M. et al. Biodiesel Made with Sugar Catalyst. Nature 438, 178 (2005).
Okamura, M. et al. Acid-Catalyzed Reactions on Flexible Polycyclic Aromatic Carbon in Amorphous Carbon. Chem. Mater. 18, 3039–3045 (2006).
Predoi, D., Kuncser, V., Filoti, g. & Schinteie, G. Magnetic Properties of γ-Fe2O3 Nanoparticles. J. Optoelectron. Adv. Mater. 5, 211–216 (2003).
Sing, K. S. W. et al. Reporting Physisorption Data for Gas/Solid Systems with Special Reference to the Determination of Surface Area and Porosity. Pure & appl. Chem. 57, 603–619 (1985).
Barrett, E. P., Joyner, L. G. & Halenda, P. P. The Determination of Pore Volume and Area Distribution in Porous Substances. I. Computations from Nitrogen Isotherms. J. Am. Chem. Soc. 73, 373–380 (1951).
Wang, Y. et al. Mobile Uranium(IV)-Bearing Colloids in a Mining-impacted Wetland. Nat. Commun. 4, 2942; 10.1038/ncomms3942 (2013).
Acknowledgements
This research was supported by a JSPS KAKENHI Grant-in-Aid for Young Scientists (B), Grant Number 24750151 and JST A-STEP (Adaptable & Seamless Technology Transfer Program through Target-driven R&D, Grant Numbers AS231Z01803D and 241FT0289). This work was also supported by Motoyama Gokin Seisakusyo Co., Ltd., Okayama Prefecture Industrial Promotion Foundation and the Yakumo Foundation for Environmental Science. We thank Prof. J. Takada and Dr. M. Nakanishi, both of Okayama University, Japan, for technical assistance with Raman and VSM analyses.
Author information
Authors and Affiliations
Contributions
D.Y. conceived and supervised the project. D.Y., K.F., M.T. and K.W. carried out experiments. D.Y., K.F., M.T. and K.W. discussed the obtained results in detail. D.Y. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Supplementary Information
Supplementary information A Magnetic Carbon Sorbent for Radioactive Material of Fukushima Nuclear Accident
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Yamaguchi, D., Furukawa, K., Takasuga, M. et al. A Magnetic Carbon Sorbent for Radioactive Material from the Fukushima Nuclear Accident. Sci Rep 4, 6053 (2014). https://doi.org/10.1038/srep06053
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06053
This article is cited by
-
Remediation of environmental toxicants using carbonaceous materials: opportunity and challenges
Environmental Science and Pollution Research (2023)
-
Comparison study of ruthenium sorption on Fe3O4 and Fe3O4@MnO2 in hydrochloric and nitric acids
Journal of Radioanalytical and Nuclear Chemistry (2021)
-
Immobilization of Bacterial Cells in Hydrogels Prepared by Gamma Irradiation for Bioremoval of Strontium Ions
Water, Air, & Soil Pollution (2020)
-
Removal of heavy metals and pollutants by membrane adsorption techniques
Applied Water Science (2018)
-
Hydrolysis of Cellulose by a Mesoporous Carbon-Fe2(SO4)3/γ-Fe2O3 Nanoparticle-Based Solid Acid Catalyst
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
