
Abstract
Repeated stress causes an energy-compromised status in the brain, with a decrease in glucose utilization by the brain cells, which might account for excitotoxicity processes seen in this condition. In fact, brain glucose metabolism mechanisms are impaired in some neurodegenerative disorders, including stress-related neuropsychopathologies. More recently, it has been demonstrated that some synthetic peroxisome proliferator-activated receptor gamma (PPARγ) agonists increase glucose utilization in rat cortical slices and astrocytes, as well as inhibit brain oxidative damage after repeated stress, which add support for considering these drugs as potential neuroprotective agents. To assess if stress causes glucose utilization impairment in the brain and to study the mechanisms by which this effect is achieved, young-adult male Wistar rats (control and immobilized for 6 h during 7 or 14 consecutive days, S7, S14) were i.p. injected with the natural ligand 15-deoxy-Δ-12,14-prostaglandin J2 (PGJ2, 120 μg/kg) or the high-affinity ligand rosiglitazone (RG, 3 mg/kg) at the onset of stress. Repeated immobilization during 1 or 2 weeks produces a decrease in brain cortical synaptosomal glucose uptake, and this effect was prevented by treatment with both natural and synthetic PPARγ ligands by restoring protein expression of the neuronal glucose transporter, GLUT-3 in membrane fractions. On the other hand, treatment with PPARγ ligands prevents stress-induced ATP loss in rat brain. Finally, repeated immobilization stress also produces a decrease in brain cortical synaptosomal glutamate uptake, and this effect was prevented by treatment with PPARγ ligands by restoring synaptosomal protein expression of the glial glutamate transporter, EAAT2. In summary, our results demonstrate that 15d-PGJ2 and the thiazolidinedione rosiglitazone increase neuronal glucose metabolism, restore brain ATP levels and prevent the impairment in glutamate uptake mechanisms induced by exposure to stress, suggesting that this class of drugs may be therapeutically useful in conditions in which brain glucose levels or availability are limited after exposure to stress.
Similar content being viewed by others


INTRODUCTION
Exposure to stress is acknowledged to be involved in a wide range of physiological and psychopathological processes. In the brain, most of the deleterious consequences after stress (molecular, cellular and morphological changes, and even mood disorders) are attributed to supraphysiological effects of glucocorticoids (reviewed in Sapolsky (2000)) as a consequence of a compromised metabolic capacity of the brain induced by overexposure to these stress hormones (reviewed in McEwen (2003), Raison and Miller (2003)).
It is well known that energy consumption in the mammalian brain is supplied mainly by the oxidation of glucose. During the physiological activation of the brain areas there is a correlative increase in local glucose uptake (Sokoloff, 1999). However, several studies have found that in stressful situations, supraphysiological levels of corticosterone have inhibitory effects on glucose transport and metabolism in several brain regions in rats (Landgraf et al, 1978; Virgin et al, 1991) and also in humans (De León et al, 1997). In peripheral tissues, glucocorticoids released by stress act by decreasing glucose transport and metabolism and increasing serum glucose levels (Munck, 1971).
This decrease in brain glucose transport by the excess of glucocorticoids during stress exposure accounts for the decrease in ATP production observed in hippocampal neurons and glia (Tombaugh and Sapolsky, 1992; Lawrence and Sapolsky, 1994). The consequence is an increased vulnerability to excitotoxicity (ie, calcium overload, reduction in the reuptake of glutamate capacity of neurons) (Novelli et al, 1988; Cheng and Mattson, 1992; Joëls and Vreugdenhil, 1998) and less ability to afford the costly task of managing the consequences of an excitotoxic or metabolic insult (Yusim et al, 2000). This impairment in the brain glucose uptake, ATP loss and excitotoxicity has been seen not only after stress or glucocorticoids, but also as an early step before neuronal degeneration takes place, such as in Alzheimer disease (Hoyer et al, 1988).
Together with these deleterious effects of stress on energy production and excitotoxic processes in the brain, the oxidative/nitrosative and neuroinflammatory consequences of stress in the central nervous system are now receiving great attention (Elenkov and Chrousos, 2002). Indeed, it is now well accepted that inflammatory responses in the brain contribute to the cell death and damage during neurological and neuropsychiatric diseases related to stress exposure (neurodegenerative diseases, depression, post-traumatic stress disorder and schizophrenia) (reviewed in McLeod et al (2001)).
Recently, it has been shown that the peroxisome proliferator-activated receptors (PPARs) are involved in the regulation of inflammatory responses (Murphy and Holder, 2000; Chawla et al, 2001). The activation of one of their three major subtypes—PPARγ—contributes by reducing the secretion of proinflammatory cytokines and neurotoxic substances in brain cells (Petrova et al, 1999), that is downregulating inducible nitric-oxide synthase expression, reducing cell death in in vitro (Heneka et al, 1999) and in vivo (Heneka et al, 2000) brain experiments. The possible clinical use of PPARγ agonists in neurological diseases has been demonstrated in experimental models, and this has led to the possibility that PPARγ agonists provide protection in neurodegenerative diseases, such as Alzheimer's disease (Heneka et al, 2001), stroke (Uryu et al, 2002), Parkinson's disease (Breidert et al, 2002), and multiple sclerosis (Niino et al, 2001).
Previous studies from our laboratory have demonstrated that both synthetic and natural PPARγ ligands prevent inflammatory and oxidative/nitrosative consequences of stress exposure in the central nervous system of rats subjected to immobilization stress (García-Bueno, 2005a, 2005b). The mechanisms by which these compounds prevent these consequences include inhibition of stress-induced increase in inducible nitric oxide synthase activity, nuclear factor κB blockade (by preventing stress-induced IκBα decrease) and inhibition of TNFα release in stressed animals. Therefore, we are particularly interested in the search of other mechanisms of protection due to PPARγ activation in the stressed brain.
Along with the interference with expression and release of mediators of insulin resistance, the improvement in glucose uptake by cells in patients with uncomplicated type 2 diabetes has been demonstrated as the main antidiabetic mechanism of action of some of the clinically available PPARγ agonists thiazolidinediones (TZDs) (reviewed in Stumvoll and Haring (2002)). Indeed, studies carried out in adipose tissue, liver, and muscle (Chawla et al, 1994; Norris et al, 2003) showed that in periphery, PPARγ regulates glucose metabolism by increasing glucose uptake through facilitative glucose transporter proteins (GLUTs). More recently, it has been demonstrated that some synthetic PPARγ agonists increase glucose utilization in rat cortical slices as well as astroglial glucose metabolism (Dello Russo et al, 2003), adding further support for considering these drugs as potential neuroprotective agents.
As brain glucose metabolism could be adversely affected in some neurodegenerative disorders, including stress-related neuropsychopathologies (reviewed in Peppard et al (1990), Hoyer (2000), Weinstock and Shoham (2004), Peters et al (2004)), we hypothesized that one mechanism of PPARγ agonists' protection during stress exposure could be related to an increase in the cerebral metabolism of glucose. The delivery of glucose within the brain is mediated primarily by GLUT-1, present in the blood–brain barrier and in astrocytes and by GLUT-3 present in neurons (Vannucci et al, 1997; Pellerin, 2005). As 85% of the energy expenditure of the brain occurs in neurons (Attwell and Laughlin, 2001), and glucose is almost the only fuel used by the organ, we tested the possibility that the preventive effects of PPARγ agonists are through glucose transporters, possibly through the neuronal component, GLUT-3. Furthermore, we examined the possibility that PPARγ agonists prevent stress-induced ATP loss and excitotoxicity in an animal model that has been reported to cause accumulation of oxidative mediators (reviewed in Madrigal et al (2004)) to explore the mechanisms implicated. To test this hypothesis, we examined the effects of two PPARγ agonists: the natural ligand 15-deoxy-Δ-12,14-prostaglandin J2 (PGJ2, Kliewer et al, 2001) and one of the synthetic TZDs, rosiglitazone (RG, Houseknecht et al, 2002).
MATERIALS AND METHODS
Animals
Adult male Wistar rats (Han:Wist, ANUC) weighing 225–250 g were used. All experimental protocols adhered to the guidelines of the Animal Welfare Committee of the Universidad Complutense following European legislation (2003/65/EC). The rats were housed individually under standard conditions of temperature and humidity and a 12-h light/dark cycle (lights on at 0800) with free access to food and water. All animals were maintained under constant conditions for 4 days prior to stress.
Restraint Stress
Rats were exposed to 6 h stress between 0900 and 1500 in the animal homeroom. The restraint was performed using a plastic rodent restrainer that allowed for a close fit to rats for 7 or 14 days (Leza et al, 1998) in their home cages. Control animals were not subjected to stress, but were handled at 0900 for a few seconds. Animals were killed immediately after restraint (still in the restrainer) using sodium pentobarbital. Blood for plasma determinations was collected by cardiac puncture and anti-coagulated in the presence of tri-sodium citrate (3.15% w:v, 1 vol citrate per 9 vol blood). After decapitation, the brain was removed from the skull and one cortical area was excised from the brain and frozen at −80°C until assayed. The other cortex was processed to obtain synaptosomes (as described below).
Plasma Corticosterone
Plasma was obtained from blood samples by centrifuging the sample at 1000g for 15 min immediately after stress on day 7 or 14. All plasma samples were stored at −20°C before assay by using a commercially available kit by RIA of 125I-labeled rat corticosterone (DPC, Los Angeles, CA, USA). A gamma counter was used to measure radioactivity of the samples. The values obtained in control animals (208.43±15.09 ng/ml) match with the kit manufacturer's expected values in adult male Wistar rats at the time of blood extraction (≈1500 h).
Preparation of Synaptosomes
After decapitation, half the forebrain was dissected on ice. All subsequent steps were performed at 4°C. Cortical tissue was immediately homogenized in 25 vol of 0.32 M sucrose in a glass homogenizer fitted with a Teflon pestle. The homogenate was centrifuged at 200g for 10 min, and the supernatant was then collected and centrifuged at 20 000g for 20 min. The pellet was resuspended in 0.32 M sucrose and centrifuged at 20 000g for 20 min. The crude synaptosomal pellet was finally resuspended in Locke solution (NaCl 154 mM, KCl 5.6 mM, CaCl2 2.3 mM, MgCl2 1 mM, NaHCO3 3.6 mM, glucose 5 mM, HEPES 5 mM, pH 7.2) without glucose for the 2-deoxy [3H] glucose transport assay and in 1 ml of 0.32 M sucrose for the glutamate transport assay. Synaptosomal preparations, consisting of pre- and post-synaptic elements of neurons, and associated astrocytic end feet, have provided insights into regulation of a variety of synaptic functions including neurotransmitter release and reuptake, energy metabolism and ion transport systems in physiological, neurological, and neurodegenerative disorders (reviewed in Begley et al (1998)).
2-Deoxy [3H] Glucose Uptake by Synaptosomes
2-Deoxy [3H] glucose uptake in synaptosomes was measured according to the procedure described previously (Keller et al, 1997) with some modifications. Briefly, synaptosomes (200 μg/tube) were washed twice in glucose-free Locke's solution, and the assay was started by the addition of 1 μCi of 2-deoxy [3H] glucose. The incubation lasted 7 min at 37°C in a shaking bath and was stopped by pelleting the synaptosomes washing twice with cold glucose-free Locke's solution and lysing the synaptosomes in 200 μl of 1% sodium dodecyl sulfate (SDS)/PBS solution. The 3H-bound radioactivity was measured using a liquid scintillation counter (Beckman LS-6500).
Brain ATP Levels
ATP levels were determined with a firefly luciferin-luciferase assay commercial kit (ATP Bioluminescence Assay Kit HSII, Roche, Barcelona, Spain) using a Fluoroskan Ascent FL microplate reader (Labsystems, Helsinki, Finland). Data are expressed as percentage of control levels. Basal ATP levels are expressed in μmol/g (modified from Hurtado et al, 2003).
[3H]Glutamate Uptake by Synaptosomes
Sodium-dependent glutamate uptake in synaptosomes was measured according to the procedure previously described by Robinson et al (1991) with some modifications. In brief, 25-μl aliquots of synaptosomes were added to 250 μl of incubation buffer (5 mM Tris, 10 mM HEPES, 2.5 mM KCl, 1.4 M NaCl, 1.2 mM CaCl2, 1.2 mM MgCl2, 1.2 mM KH2PO4, and 10 mM dextrose, pH 7.4) containing L-[3H]glutamic acid 0.125 μM (1 mCi/ml; Amersham Biosciences Europe GmbH, Friburg, Germany) and incubated for 3 min at 37°C in a shaking bath. The reaction was terminated using 1 ml of ice-cold choline buffer (incubation buffer in which an equimolar concentration of choline chloride was substituted for NaCl), and the samples were centrifuged at 10 000g for 2 min to recover synaptosomes. The 3H-bound radioactivity was measured using a liquid scintillation counter.
Western Blot Analysis
Succesful synaptosomal preparations were verified by determining expression of membrane associated protein synaptophysin (Sigma, 1:5000) and the absence of the cytoplasmatic protein B-tubulin (Sigma, 1:1000) (Reagan et al, 2000). Synaptophysin was used as loading control. To determine the levels of these proteins and the facilitative glucose transporter proteins GLUT-1, GLUT-3, and the high-affinity sodium-dependent glutamate transporters EAAT-2 and EAAT-3, proteins present in synaptosomes were loaded (20 μg) and size-separated in 10% SDS-polyacrylamide gel electrophoresis (90 V). After blotting onto a polyvinylidene difluoride membrane (Millipore, Bedford, MA, USA), were incubated with specifics rabbit polyclonal GLUT-1 (Chemicon international, 1:3000), GLUT-3 (Chemicon international, 1:5000) EAAT-3 (Santa Cruz, 1:500) and mouse monoclonal EAAT-2 (Transduction Labs, 1:1000) antibodies. Proteins recognized by the antibody were visualized on X-ray film by chemiluminiscence (ECL) following the manufacturer's instructions (Amersham lbérica). Autorradiographs were quantified by densitometry (Scion Image, Scion Corp., Frederick, MD, USA), and several time expositions were analyzed to ensure the linearity of the band intensities. Data are presented as arbitrary units (AU) of optical density.
Pharmacological Tools
Various groups of animals were i.p. injected with rosiglitazone (RG) maleate (3 mg/kg; Alexis, San Diego, CA), a high-affinity synthetic PPARγ receptor ligand (reviewed in Berger and Moller 2002), and 120 μg/kg of 15d-PGJ2 (Cayman Chem., Ann Arbor, MI, USA). RG was dissolved in saline and 15d-PGJ2 in DMSO (10%). None of the parameters studied were modified in vehicle-treated rats when compared with noninjected animals. The drugs were injected at the onset of each daily stress session. Animals receiving vehicles at the onset of stress were used as the control group in the Results section and in figures. Each experimental group contained at least eight animals.
Protein Assay
Proteins were measured using bicinchoninic acid (Hill and Straka, 1988).
Chemicals and Statistical Analyses
Unless otherwise stated, the chemicals were from Sigma Spain, Madrid. Data in text and figures are expressed as mean±SEM of the indicated number of experiments. Multiple comparisons were analyzed by the ANOVA and Newman–Keuls test, and P<0.05 was considered as statistically significant.
RESULTS
Effects of Rosiglitazone and PGJ2 on Stress-Induced Decrease in Synaptosomal Glucose Uptake and Expression of Glucose Transporters
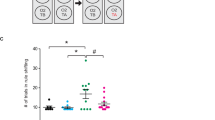
We tested the effect of rosiglitazone (RG) and PGJ2 on synaptosomal glucose uptake. Control, nonstressed rats were injected to either RG or PGJ2 for 7 or 14 days (3 mg/kg/day and 120 μg/kg/day, respectively). Cerebral glucose uptake was not modified by the compounds in control rats at the tested doses (not shown, n=4). Exposure to repeated stress (S7 and S14) leads to a marked decrease in glucose uptake (S7:60±6%; S14:68±8% vs control, 5.9±0.5 cpm/min/mg protein, both P<0.05 Figure 1). Treatment with RG and PGJ2 prevented this stress-induced impairment in glucose uptake function at both times studied (S7+RG:107±11%; S7+PGJ2:135±17%, both P<0.05 vs control; S14+RG:94±4%; S14+PGJ2:96±6%, both P<0.05 vs control) (Figure 1).

Effect of repeated restraint exposure on glucose uptake in synaptosomes of control (nonstressed) and stressed rats during 6 h for 7 days (S7) or 14 days (S14) receiving vehicle (SS, see Materials and methods for details), rosiglitazone (RG; 3 mg/kg) or 15d-PGJ2 (PGJ2 120 μg/kg), respectively at the onset of restraint. The data represent the mean SEM of eight rats. *P<0.05 vs control; #P<0.05 vs correspondent SS (S7 or S14) (Newman–Keuls test).
We also assessed the possibility that these effects might be due to expressional changes in glucose transporters. Successful isolation of cerebral membranes fraction was verified by immunoblot analysis showing the presence of the membrane protein associated to synaptic vesicles synaptophysin (Syn, MW 38 kDa), as well as the absence of the cytoplasmic protein B-tubulin (B-tub, MW 55 kDa) (Figure 2a). Western blot analysis revealed that stress reduced the expression of GLUT-3 (Figure 2b), but not the astrocytic form of GLUT-1 (45 kDa) (Figure 2c) and that treatment with RG and PGJ2 prevented this stress-induced reduction in GLUT-3 expression at both times studied (Figure 2b). The blood brain barrier form of GLUT-1 (55 kDa) was not found in the samples used in this study (not shown).

(a) Detection of synaptophysin (Syn) protein presence and absence of β-tubulin (β-tub) in synaptosomal forebrain fractions by Western Blot. (b) Western blot analysis of neuronal GLUT-3 in synaptosomal fractions from forebrain of control and stressed (S7 or S14) rats treated with saline (SS) and with rosiglitazone (3 mg/kg) (RG) or 15d-PGJ2 (120 μg/kg) (PGJ2), respectively at the onset of stress and laser densitometric analysis of the bands (arbitrary units, AU). The data are representative of samples of six different animals. *P<0.05 vs control; #P<0.05 vs S7 or S14 (Newman–Keuls test). (c) Western blot detection of astrocytic GLUT-1 (45 kDa) protein and laser densitometric analysis of the band in synaptosomes of control and stressed rats during 6 h for 7 (S7) or 14 (S14) days with or without rosiglitazone (3 mg/kg) (RG) or 15d-PGJ2 (120 μg/kg) (PGJ2), respectively at the beginning of restraint session. Western data are representative of samples of six different animals.
Effects of RG and PGJ2 on Stress-Induced Decrease in Brain ATP Levels
Exposure to repeated stress (S7 and S14) lead to a marked decrease in brain ATP levels (S7:52±6.2%; S14:63±5% vs control, 1.40±0.2 nmol/mg protein, both P<0.05, Figure 3). Treatment with RG and PGJ2 prevented this stress-induced decrease in ATP at both times studied (S7+RG:84±9; S7+PGJ2:88±8, both P<0.05 vs control; S14+RG:95±8; S14+PGJ2:92±7, both P<0.05 vs control) (Figure 3). Control, nonstressed rats were injected with either RG or PGJ2 for 7 or 14 days (3 mg/kg/day or 120 μg/kg/day, respectively), but ATP levels were not modified with the compounds at the tested doses (not shown, n=4).

ATP content in brain cortex of control and stressed rats during 6 h for 7 days (S7) or 14 days (S14) receiving vehicle (SS), rosiglitazone (RS; 3 mg/kg) or 15d-PGJ2 (120 μg/kg) (PGJ2) respectively at the onset of stress protocol. The data represent the mean±SEM of eight rats. *P<0.05 vs control (Newman–Keuls test). #P<0.05 vs SS.
Effects of RG and PGJ2 on Stress-Induced Decrease in Synaptosomal Glutamate Uptake and in the Expression of Glutamate Transporters
We tested the effect of rosiglitazone and PGJ2 on synaptosomal glutamate uptake. Control, nonstressed rats were injected with either RG or PGJ2 for 7 or 14 days (3 mg/kg/day and 120 μg/kg/day, respectively). Cerebral glutamate uptake was not modified with the compounds at the tested doses (not shown, n=4). Exposure to repeated stress (S7 and S14) leads to a marked decrease in glutamate uptake (S7:45±7; S14:53±4% vs control, 4±0.0015 nmol/min/mg prot. both P<0.05, Figure 4). Treatment with RG and PGJ2 prevented this stress-induced impairment in glutamate uptake function at both times studied (S7+RG:82±15; S7+PGJ2:125±20, both P<0.05 vs control; S14+RG:68±8; S14+PGJ2:78±10, both P<0.05 vs control) (Figure 4).

Effect of restraint stress on glutamate uptake in forebrain synaptosomes of control (nonstressed) and stressed rats for 6 h during 7 (S7) or 14 (S14) days receiving vehicle (SS, see Materials and methods for details), rosiglitazone (RG; 3 mg/kg) or 15d-PGJ2 (PGJ2 120 μg/kg), respectively at the onset of restraint. The data represent the mean SEM of eight rats. *P<0.05 vs control; #P<0.05 vs S7 or S14 (Newman–Keuls test).
We also assessed the possibility that these effects might be due to expressional changes in glutamate transporters. Western blot analysis revealed that stress reduced the expression of EAAT2 (Figure 5a), but not EAAT1 (not shown) or EAAT3 (Figure 5b) and that treatment with RG and PGJ2 prevented this stress-induced reduction in EAAT-2 expression at both times studied (Figure 5a).

(a) Western Blot detection of astrocytic EAAT-2 protein and laser densitometric analysis of the band (arbitrary units, AU) in synaptosomes of control and stressed rats during 6 h for 7 (S7) or 14 (S14) days with or without rosiglitazone (3 mg/kg) (RG) or 15d-PGJ2 (120 μg/kg) (PGJ2), respectively at the onset of stress protocol. Western data are representative of samples of six different animals. *P<0.05 vs control; #P<0.05 vs S7 or S14 (Newman-Keuls test) (SS: vehicle, see Materials and methods for details). (b) Western blot analysis of neuronal EAAT-3 protein and laser densitometric analysis of the band (AU) in synaptosomal fraction of control and stressed rats during 6 h for 7 (S7) or 14 (S14) days with or without rosiglitazone (3 mg/kg) (RG) or 15d-PGJ2 (120 μg/kg) (PGJ2), respectively at the beginning of restraint session. Western data are representative of samples of six different animals (SS: vehicle see Materials and methods for details).
Absence of Effects of RG and PGJ2 on Stress-Induced Increase in Plasma Corticosterone Levels
To test the possibility that the effects of selective PPARγ activation were related to interference on the general response to stress, we determined plasma corticosterone levels in all of the experimental groups studied. The effects elicited by the PPARγ ligands after stress in the brain are independent of the systemic stress response, as plasma corticosterone levels (at 1500 h) were not modified in rats injected at the onset of stress: control, saline-injected at 0900 h:208.43±15.09 ng/ml; control rats receiving RG 3 mg/kg:221.92±65.03; receiving PGJ2 120 μg/kg:191.03±39.99 ng/ml, both P>0.05 vs control, saline; stress 7d:325.55±16.02 ng/ml, P<0.05 vs control; stress 7d plus RG:297.12±20.02 ng/ml, P>0.05 vs stress 7d; stress 7d plus PGJ2:302.55±25.71 ng/ml, P>0.05 vs stress 7d. Finally, stress 14d:346.95±45.11 ng/ml, P<0.05 vs control; stress 14d plus RG:310.10±32.22 ng/ml, P>0.05 vs stress 14d; stress 14d plus PGJ2:325.62±48.84 ng/ml, P>0.05 vs stress 14d.
DISCUSSION
All of the findings presented here indicate that repeated stress causes an energy-compromised status in the brain which might account for excitotoxicity seen in this condition and that treatment with PPARγ ligands prevents all these changes. In particular, the reported results demonstrate that repeated immobilization stress during 1 or 2 weeks produces a decrease in brain cortical synaptosomal glucose uptake, this effect being prevented by treatment with both natural and synthetic PPARγ ligands by restoring protein expression of the neuronal glucose transporter, GLUT-3, in membrane fraction. Besides, brain ATP production is decreased in rats exposed to stress, and treatment with PPARγ ligands prevents stress-induced ATP loss. Finally, repeated immobilization stress also produces a decrease in brain cortical synaptosomal glutamate uptake, this effect being prevented by treatment with PPARγ ligands by restoring protein expression of the glial glutamate transporter, EAAT2 in synaptosomes.
Glucose transport from blood into the brain is mediated by a facilitated diffusion-type transport system, that is members of the GLUT supergene family of integral membrane proteins (Maher et al, 1994; Vannucci et al, 1997; Shepherd and Kahn, 1999). Tissue-specific expression of one or more members determines the rate of glucose entry into the cell. In brain, GLUT-1 and GLUT-3 seem to be the most important. GLUT-1, which is expressed and localized at the endothelial cells of the blood-brain barrier (in a glycosylated form with 55 kDa), is responsible for the majority of glucose uptake and utilization in the brain (Duelli and Kuschinsky, 2001) and takes part in the first step in the transport of glucose from the blood into the brain (Pardridge et al, 1990; Boado and Pardridge, 1990). On the membrane of end feet around blood vessels, astrocytes express a specific form of GLUT-1, with 45 kDa (reviewed in Pellerin (2005)). The next step of glucose transport from extracellular space into neuronal cells is taken by GLUT-3, localized at the neuronal cell membrane (Maher, 1995). GLUT-3 possesses higher affinity (lower kilometers) for glucose than GLUT-1 (reviewed in Bolaños et al (2004)). Taking into account the high affinity and dependence of neurons for glucose, it has been postulated that the activity of this transporter would play a beneficial role during glucose deprivation, hypoxic episodes or other metabolic compromising situations (Burant and Bell, 1992; Gerhart et al, 1992; Fattoretti et al, 2001; Burkhalter et al, 2003). Thus, GLUT-3 activity affords neuroprotection (Hara et al, 1989; Lee and Bondy, 1993; Urabe et al, 1996; Uehara et al, 1997), and its malfunctioning is related with neuronal damage (reviewed in McEwen and Reagan (2004)). We have shown that after repeated stress, at the stress intensity and duration evaluated in this article, the neuronal transporter GLUT-3 membrane expression results clearly affected supporting the preventive effects demonstrated of the PPARγ agonists used in this particular experimental setting (García-Bueno et al, 2005a, 2005b).
The results of the present study indicate that astrocytic GLUT-1 expression is not modified after 1 or 2 weeks of repeated stress. Membrane preparations such as synaptosomes are free from BBB cells. Thus, GLUT-1 should come from astrocytes, as verified by identification of the astrocytic form (45 kDa), but not the 55 kDa form, in the samples used in this study. The presence of this specific isoform of GLUT-1 has been previously demonstrated in rat brain synaptosomal preparations by Bhattacharyya and Brodsky (1988).
In our study, plasma corticosterone levels are beyond physiological levels, which is in agreement with the fact that glucocorticoids dose-dependently inhibit brain glucose uptake when they are present in the brain at concentrations above the high physiological range in in vitro (Virgin et al, 1991) and in vivo (Doyle et al, 1994). Interestingly, similar GC treatment caused a decrease in the affinity of glutamate uptake by astrocytes. Studies have shown that in hippocampal cell cultures glucocorticoids significantly inhibit glucose transport and glutamate uptake by both neurons and astrocytes (Horner et al, 1990; Virgin et al, 1991). This latter observation suggests that GCs might impair the ability of astrocytes to aid neurons (ie, by impairing their ability to remove damaging glutamate from the synapse), which may be the case of our model.
Two possible mechanisms have been proposed to explain the stress-induced decrease in GLUT-3 expression: modification in binding to plasma membrane (Horner et al, 1987) or oxidative attack by mediators released during stress (Reagan et al, 2000). The first possible mechanism of GLUTs regulation is mediated by translocation from the plasma membrane to intracellular sites, as demonstrated previously after GC treatment (Horner et al, 1987). Many studies have shown that fusion of GLUT-3 vesicles with the plasma membrane increases glucose uptake (Uemura and West Greenlee, 2001). The fact that the effects observed here were seen on an enriched membrane preparation (synaptosomes) suggests that this may be the case in stress-induced decrease in glucose uptake. The second possible mechanism for the inhibitory stress effects on glucose uptake is the oxidative damage of GLUT-3 evidenced by conjugation of 4-hydroxynonenal (HNE) (Mark et al, 1997) or other lipid peroxidation products released in brain in the same stress model used in this paper (daily immobilization for 6 h during 7 days) (Reagan et al, 2000). This has also been seen in other settings such as cultured hippocampal neurons exposed to Abeta (increased HNE production and conjugation to GLUT-3) (Mark et al, 1997). In the stress model used here, the production of oxidative and nitrosative mediators and the accumulation of lipid peroxidation markers in the brain have been widely documented (Liu et al, 1996; Madrigal et al, 2001).
The reported decrease in glucose uptake mediated by stress or by high levels of GCs could place neurons in an energy-compromised environment, which could detrimentally affect neuronal responsiveness to pathophysiological events. In fact, glucose transport impairment precedes ATP depletion in brain, increasing neuronal vulnerability to excitotoxicity by compromising function of ion-motive ATPases (Mark et al, 1995), as it has been seen to occur in some neurodegenerative processes such Ab-induced toxicity, schizophrenia and others (Novelli et al, 1988; Kalaria and Harik, 1989; Sims, 1990; Mark et al, 1997; McDermott and De Silva, 2005) in which the reduction in glucose uptake occurs as an early step in the disease process prior to neuronal degeneration (Pettegrew et al, 1994; Reiman et al, 1996). On the other hand, GCs promote reduction in ATP levels (Tombaugh and Sapolsky, 1992; Lawrence and Sapolsky, 1994). One of the possible ATP-dependent mechanisms compromised by stress are glutamate transporters.
A likely functional implication of the stress effects on cerebral glucose transport is an impairment of glutamate uptake (Virgin et al, 1991). Indeed, stress has been shown to increase extracellular glutamate concentrations in many brain areas (Lowy et al, 1993; Moghaddam et al, 1994; Stein-Behrens et al, 1994), an effect that is proposed to result from compromised activity of the energy-dependent excitatory amino acid transporters. On the other hand, it has recently been shown that glutamate release is mainly due to reversed operation of neuronal glutamate transporters in processes such as brain ischemia and others (Warner et al, 1996; Jabaudon et al, 2000; Rossi et al, 2000). Thus, in stress, this is one among the various mechanisms, which, alone or combined, may be responsible for glutamate release (Lawrence and Sapolsky, 1994) although we have not seen modifications in glutamate uptake in neurons (EAAT-3). The effects elicited by the type of stress used in this study seem to be restricted to astrocytic mechanisms: a large percentage of the neurotransmitter is removed from the synapse via this route. Astrocyte glutamate uptake is a high affinity process regulated by EAAT-2 (Hertz et al, 1983), and the fact that stress decreases EAAT-2 expression observed here indicates a specific damaging effect at this level. Thus, the excess of extracellular glutamate could be a feedback mechanism, as has been proposed between stress, glucose and glutamate in the brain, as glutamate stimulates the HPA axis promoting a continuous circle (Gabr et al, 1995). Interestingly, this change seems to be time-dependent and region-specific: after repeated stress (21–40 days), an increase in EAAT-2 has been found in hippocampus, which has been proposed as a counter-regulatory mechanism (Reagan et al, 2004; Fontella et al, 2004).
The demonstration of the PPARγ agonists effects used in this study, PGJ2 and rosiglitazone (RG), after stress has important implications for possible pharmacological management of stress-related neuropsychopathologies, in which energy status may be affected. Rosiglitazone is a member of the peroxisome proliferator activated receptor gamma (PPARγ) agonists of the thiazolidinedione family, which are widely used as antidiabetic agents. Rosiglitazone therapy improves insulin sensitivity and glucose uptake in patients with uncomplicated type 2 diabetes. In addition to the effects on glucose metabolism, both TZDs and natural PPARγ ligands have effects on lipid metabolism, inflammatory responses, and cellular proliferation (reviewed in Kostadinova et al (2005)). Finally, several studies have demonstrated an enhancement of glucose uptake and glucose transporter expression in adipocytes and myocyte membranes treated with TZDs (Ciaraldi and Henry, 1997).
Treatment with these drugs is effective in overcoming stress inhibition of neuronal glucose uptake, increasing glucose utilization and GLUT-3 expression. One of the possible mechanisms is the prevention of cerebral GLUT-3 oxidation, which occurs in stress (Reagan et al, 2000) as it has been demonstrated to occur in stressed brain (García-Bueno et al, 2005a, 2005b).
In conclusion, our results demonstrate that TZDs can increase the brain glucose metabolism, suggesting that this class of drugs may be therapeutically useful in conditions in which brain glucose levels or availability are limited.
Besides, rosiglitazone has been found to prevent the loss of ATP in in vitro hearts (Sidell et al, 2002). Our findings open up a line of research directed toward the elucidation of other roles of thiazolidindiones and 15d-PGJ2 by affecting the ATP levels in brain cells, which may have important implications after stress at the level of glutamate uptake and release.
To our knowledge, this is the first work that demonstrates the antiexcitotoxic effect of PPARγ agonists at the level of the expression and activity of the EAATs family. Recently, other authors have showed neuroprotective antiexcitotoxic properties of these compounds blocking the NMDA-receptor-mediated Ca2+ entry (Zhao et al, 2006), preventing oxidative stress (Aoun et al, 2003) or regulating the PI3-kinase; (Nishijima et al, 2001), effects that took place in our model of stress.
In summary, our present findings show that these drugs exert direct actions on cerebral glucose and glutamate metabolism added to its known antiinflamatory/antioxidant effects, adding new therapeutic implications in the management of patients at risk of stressful events.
References
Aoun P, Watson DG, Simpkins JW (2003). Neuroprotective effects of PPARgamma agonists against oxidative insults in HT-cells. Eur J Pharmacol 472: 65–71.
Attwell D, Laughlin SB (2001). An energy budget for signaling in the grey matter of the brain. J Cereb Blood Flow Metab 21: 1133–1145.
Begley JG, Butterfield DA, Keller JN, Koppal T, Drake J, Mattson MP (1998). Cryopreservation of rat cortical synaptosomes and analysis of glucose and glutamate transporter activities, and mitochondrial function. Brain Res Brain Res Protoc 3: 76–82.
Berger J, Moller DE (2002). The mechanisms of action of PPARs. Annu Rev Med 53: 409–435.
Bhattacharyya MV, Brodsky JL (1988). Characterization of the glucose transporter from rat brain synaptosomes. Biochem Biophys Res Commun 15: 685–691.
Boado RJ, Pardridge WM (1990). The brain-type glucose transporter mRNA is specifically expressed at the blood–brain barrier. Biochem Biophys Res Commun 166: 174–179.
Bolaños JP, Cidad P, Garcia-Nogales P, Delgado-Esteban M, Fernandez E, Almeida A (2004). Regulation of glucose metabolism by nitrosative stress in neural cells. Mol Aspects Med 25: 61–73.
Breidert T, Callebert J, Heneka MT, Landreth G, Launay JM, Hirsch EC (2002). Protective action of the peroxisome proliferator-activated receptor-gamma agonist pioglitazone in a mouse model of Parkinson's disease. J Neurochem 82: 615–624.
Burant CF, Bell GI (1992). Mammalian facilitative glucose transporters: evidence for similar substrate recognition sites in functionally monomeric proteins. Biochemistry 31: 10414–10420.
Burkhalter J, Fiumelli H, Allaman I, Chatton JY, Martin JL (2003). Brain-derived neurotrophic factor stimulates energy metabolism in developing cortical neurons. J Neurosci 23: 8212–8220.
Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM (2001). PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med 7: 48–52.
Chawla A, Schwarz EJ, Dimaculangan DD, Lazar MA (1994). Peroxisome proliferator-activated receptor (PPAR) gamma: adipose-predominant expression and induction early in adipocyte differentiation. Endocrinology 135: 798–800.
Cheng B, Mattson MP (1992). Glucose deprivation elicits neurofibrillary tangle-like antigenic changes in hippocampal neurons: prevention by NGF and bFGF. Exp Neurol 117: 114–123.
Ciaraldi T, Henry RR (1997). Thiazolidinediones and their effects on glucose transporters. Eur J Endocrinol 137: 610–612.
De Leon MJ, McRae T, Rusinek H, Convit A, De Santi S, Tarshish C et al (1997). Cortisol reduces hippocampal glucose metabolism in normal elderly, but not in Alzheimer's disease. J Clin Endocrinol Metab 82: 3251–3259.
Dello Russo C, Gavrilyuk V, Weinberg G, Almeida A, Bolaños JP, Palmer J et al (2003). Peroxisome proliferator-activated receptor gamma thiazolidinedione agonists increase glucose metabolism in astrocytes. J Biol Chem 278: 5828–5836.
Doyle P, Guillaume-Gentil C, Rohner-Jeanrenaud F, Jeanrenaud B (1994). Effects of corticosterone administration on local cerebral glucose utilization of rats. Brain Res 645: 225–230.
Duelli R, Kuschinsky W (2001). Brain glucose transporters: relationship to local energy demand. News Physiol Sci 16: 71–76.
Elenkov IJ, Chrousos GP (2002). Stress hormones, proinflammatory and antiinflammatory cytokines, and autoimmunity. Ann NY Acad Sci 966: 290–303.
Fattoretti P, Bertoni-Freddari C, Di Stefano G, Casoli T, Gracciotti N, Solazzi M et al (2001). Quantitative immunohistochemistry of glucose transport protein (Glut 3) expression in the rat hippocampus during aging. J Histochem Cytochem 49: 671–672.
Feinstein DL (2003). Therapeutic potential of peroxisome proliferator-activated receptor agonists for neurological disease. Diabet Technol Ther 5: 67–73.
Fontella FU, Vendite DA, Tabajara AS, Porciuncula LO, da Silva Torres IL, Jardim FM et al (2004). Repeated restraint stress alters hippocampal glutamate uptake and release in the rat. Neurochem Res 29: 1703–1709.
Gabr RW, Birkle DL, Azzaro AJ (1995). Stimulation of the amygdala by glutamate facilitates corticotropin-releasing factor release from the median eminence and activation of the hypothalamic–pituitary–adrenal axis in stressed rats. Neuroendocrinology 62: 333–339.
García-Bueno B, Madrigal JL, Lizasoain I, Moro MA, Lorenzo P, Leza JC (2005a). Peroxisome proliferator-activated receptor gamma activation decreases neuroinflammation in brain after stress in rats. Biol Psychiatry 57: 885–894.
García-Bueno B, Madrigal JL, Lizasoain I, Moro MA, Lorenzo P, Leza JC (2005b). The anti-inflammatory prostaglandin 15d-PGJ2 decreases oxidative/nitrosative mediators in brain after acute stress in rats. Psychopharmacology 180: 513–522.
Gerhart DZ, Broderius MA, Borson ND, Drewes LR (1992). Neurons and microvessels express the brain glucose transporter protein GLUT3. Proc Natl Acad Sci 89: 733–737.
Hara M, Matsuda Y, Okumura N, Hirai K, Nakagawa H (1989). Effect of glucose starvation on glucose transport in neuronal cells in primary culture from rat brain. J Neurochem 52: 909–912.
Heneka MT, Feinstein DL, Galea E, Gleichmann M, Wullner U, Klockgether T (1999). Peroxisome proliferator-activated receptor gamma agonists protect cerebellar granule cells from cytokine-induced apoptotic cell death by inhibition of inducible nitric oxide synthase. J Neuroimmunol 100: 156–168.
Heneka MT, Klockgether T, Feinstein DL (2000). Peroxisome proliferator-activated receptor-gamma ligands reduce neuronal inducible nitric oxide synthase expression and cell death in vivo. J Neurosci 20: 6862–6867.
Heneka MT, Landreth GE, Feinstein DL (2001). Role for peroxisome proliferator-activated receptor-gamma in Alzheimer's disease. Ann Neurol 49: 276.
Hertz L, Bender AS, Richardson JS (1983). Benzodiazepines and beta-adrenergic binding to primary cultures of astrocytes and neurons. Prog Neuropsychopharmacol Biol Psychiatry 7: 681–686.
Hill HD, Straka JG (1988). Protein determination using bicinchoninic acid in the presence of sulfhydryl reagents. Anal Biochem 170: 203–218.
Horner HC, Munck A, Lienhard GE (1987). Dexamethasone causes translocation of glucose transporters from the plasma membrane to an intracellular site in human fibroblasts. J Biol Chem 262: 17696–17702.
Horner HC, Packan DR, Sapolsky RM (1990). Glucocorticoids inhibit glucose transport in cultured hippocampal neurons and glia. Neuroendocrinology 52: 57–64.
Houseknecht KL, Cole BM, Steele PJ (2002). Peroxisome proliferator-activated receptor gamma (PPARgamma) and its ligands: a review. Domest Anim Endocrinol 22: 1–23.
Hoyer S (2000). Brain glucose and energy metabolism abnormalities in sporadic Alzheimer disease. Causes and consequences: an update. Exp Gerontol 35: 1363–1372.
Hoyer S, Oesterreich K, Wagner O (1988). Glucose metabolism as the site of the primary abnormality in early-onset dementia of Alzheimer type? J Neurol 235: 143–148.
Hurtado O, De Cristóbal J, Sánchez V, Lizasoain I, Cárdenas C, Pereira MP et al (2003). Inhibition of glutamate release by delaying ATP fall accounts for neuroprotective effects of antioxidants in experimental stroke. FASEB J 17: 2082–2084.
Jabaudon D, Scanziani M, Gahwiler BH, Gerber U (2000). Acute decrease in net glutamate uptake during energy deprivation. Proc Natl Acad Sci 97: 5610–5615.
Joels M, Vreugdenhil E (1998). Corticosteroids in the brain. Cellular and molecular actions. Mol Neurobiol 17: 87–108.
Kalaria RN, Harik SL (1989). Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J Neurochem 53: 1083–1088.
Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G et al (1997). Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by amyloid beta-peptide: role of the lipid peroxidation product 4-hydroxynonenal. J Neurochem 69: 273–824.
Kliewer SA, Xu HE, Lambert MH, Willson TM (2001). Peroxisome proliferator-activated receptors: from genes to physiology. Recent Prog Horm Res 56: 239–263.
Kostadinova R, Wahli W, Michalik L (2005). PPARs in diseases: control mechanisms of inflammation. Curr Med Chem 12: 2995–3009.
Landgraf R, Mitro A, Hess J (1978). Regional net uptake of 14C-glucose by rat brain under the influence of corticosterone. Endocrinol Exp 12: 119–129.
Lawrence MS, Sapolsky RM (1994). Glucocorticoids accelerate ATP loss following metabolic insults in cultured hippocampal neurons. Brain Res 646: 303–306.
Lee WH, Bondy CA (1993). Ischemic injury induces brain glucose transporter gene expression. Endocrinology 133: 2540–2544.
Leza JC, Salas E, Sawicki G, Russell JC, Radomski MW (1998). The effects of stress on homeostasis in JCR-LA-cp rats: the role of nitric oxide. J Pharmacol Exp Ther 286: 1397–1403.
Liu J, Wang X, Shigenaga MK, Yeo HC, Mori A, Ames BS (1996). Immobilization stress causes oxidative damage to lipid, protein and DNA in the brain of rats. FASEB J 10: 1532–1538.
Lowy MT, Gault L, Yamamoto BK (1993). Adrenalectomy attenuates stress-induced elevations in extracellular glutamate concentrations in the hippocampus. J Neurochem 61: 1957–1960.
Madrigal JLM, Garcia-Bueno B, Cárdenas A, Lizasoain I, Moro MA, Lorenzo P et al (2004). Oxidative/nitrosative brain damage in stress: possible target for neuropsychopharmacological drugs. Curr Med Chem-CNS Agents 4: 235–242.
Madrigal JLM, Olivenza R, Moro MA, Lizasoain I, Lorenzo P, Rodrigo J et al (2001). Glutathione depletion, lipid peroxidation and mitochondrial dysfunction are induced by chronic stress in rat brain. Neuropsychopharmacology 24: 420–429.
Maher F (1995). Immunolocalization of GLUT-1 and GLUT-3 glucose transporters in primary cultured neurons and glia. J Neurosci Res 42: 459–469.
Maher F, Vannucci SJ, Simpson IA (1994). Glucose transporter proteins in brain. FASEB J 8: 1003–1011.
Mark RJ, Hensley K, Butterfield DA, Mattson MP (1995). Amyloid beta-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci 15: 6239–6249.
Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP (1997). Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J Neurosci 17: 1046–1054.
McDermott E, de Silva P (2005). Impaired neuronal glucose uptake in pathogenesis of schizophrenia—can GLUT 1 and GLUT-3 deficits explain imaging, post-mortem and pharmacological findings? Med Hypotheses 65: 1076–1081.
McEwen BS (2003). Mood disorders and allostatic load. Biol Psychiatry 54: 200–207.
McEwen BS, Reagan LP (2004). Glucose transporter expression in the central nervous system: relationship to synaptic function. Eur J Pharmacol 490: 13–24.
McLeod TM, Lopez-Figueroa AL, Lopez-Figueroa MO (2001). Nitric oxide, stress, and depression. Psychopharmacol Bull 35: 24–41.
Moghaddam B, Bolinao ML, Stein-Behrens B, Sapolsky RM (1994). Glucocorticoids mediate the stress-induced extracellular accumulation of glutamate. Brain Res 655: 251–254.
Munck A (1971). Glucocorticoid inhibition of glucose uptake by peripheral tissues: old and new evidence, molecular mechanisms, and physiological significance. Perspect Biol Med 14: 265–269.
Murphy GJ, Holder JC (2000). PPAR-gamma agonists: therapeutic role in diabetes, inflammation and cancer. Trends Pharmacol Sci 21: 469–474.
Niino M, Iwabuchi K, Kikuchi S, Ato M, Morohashi T, Ogata A et al (2001). Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by an agonist of peroxisome proliferator-activated receptor-gamma. J Neuroimmunol 116: 40–48.
Nishijima C, Kimoto K, Arakawa Y (2001). Survival activity of troglitazone in rat motoneurones. J Neurochem 76: 383–390.
Norris AW, Chen L, Fisher SJ, Szanto I, Ristow M, Jozsi AC et al (2003). Muscle-specific PPARgamma-deficient mice develop increased adiposity and insulin resistance but respond to thiazolidinediones. J Clin Invest 112: 608–618.
Novelli A, Reilly JA, Lysko PG, Henneberry RC (1988). Glutamate becomes neurotoxic via the N-methyl-D-aspartate receptor when intracellular energy levels are reduced. Brain Res 451: 205–212.
Pardridge WM, Boado RJ, Farrell CR (1990). Brain-type glucose transporter (GLUT-1) is selectively localized to the blood-brain barrier. Studies with quantitative Western blotting and in situ hybridization. J Biol Chem 265: 18035–18040.
Pellerin L (2005). How astrocytes feed hungry neurons. Mol Neurobiol 32: 59–72.
Peppard RF, Martin WR, Clark CM, Carr GD, McGeer PL, Calne DB (1990). Cortical glucose metabolism in Parkinson's and Alzheimer's disease. J Neurosci Res 27: 561–568.
Peters A, Schweiger U, Pellerin L, Hubold C, Oltmanns KM, Conrad M et al (2004). The selfish brain: competition for energy resources. Neurosci Biobehav Rev 28: 143–180.
Petrova TV, Akama KT, Van Eldik LJ (1999). Cyclopentenone prostaglandins suppress activation of microglia: down-regulation of inducible nitric-oxide synthase by 15-deoxy-Delta12,14-prostaglandin J2. Proc Natl Acad Sci 96: 4668–4673.
Pettegrew JW, Panchalingam K, Klunk WE, McClure RJ, Muenz LR (1994). Alterations of cerebral metabolism in probable Alzheimer's disease: a preliminary study. Neurobiol Aging 15: 117–132.
Raison CL, Miller AH (2003). When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry 160: 1554–1565.
Reagan LP, Magarinos AM, Yee DK, Swzeda LI, Van Bueren A, McCall AL et al (2000). Oxidative stress and HNE conjugation of GLUT-3 are increased in the hippocampus of diabetic rats subjected to stress. Brain Res 862: 292–300.
Reagan LP, Rosell DR, Wood GE, Spedding M, Munoz C, Rothstein J et al (2004). Chronic restraint stress up-regulates GLT-1 mRNA and protein expression in the rat hippocampus: reversal by tianeptine. Proc Natl Acad Sci 101: 2179–2184.
Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S et al (1996). Preclinical evidence of Alzheimer's disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med 334: 752–758.
Robinson MB, Hunter-Ensor M, Sinor J (1991). Pharmacologically distinct sodium-dependent L-[3H]glutamate. Brain Res 544: 196–202.
Rossi DJ, Oshima T, Attwell D (2000). Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403: 316–321.
Sapolsky RM (2000). Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry 57: 925–935.
Shepherd PR, Kahn BB (1999). Glucose transporters and insulin action—implications for insulin resistance and diabetes mellitus. N Engl J Med 341: 248–257.
Sidell RJ, Cole MA, Draper NJ, Desrois M, Buckingham RE, Clarke K (2002). Thiazolidinedione treatment normalizes insulin resistance and ischemic injury in the zucker Fatty rat heart. Diabetes 51: 1110–1117.
Sims NR (1990). Altered glucose metabolism in Alzheimer's disease. Ann Neurol 27: 691–693.
Sokoloff L (1999). Energetics of functional activation in neural tissues. Neurochem Res 24: 321–329.
Stein-Behrens BA, Lin WJ, Sapolsky RM (1994). Physiological elevations of glucocorticoids potentiate glutamate accumulation in the hippocampus. J Neurochem 63: 596–602.
Stumvoll M, Haring HU (2002). Glitazones: clinical effects and molecular mechanisms. Ann Med 34: 217–224.
Tombaugh GC, Sapolsky RM (1992). Corticosterone accelerates hypoxia- and cyanide-induced ATP loss in cultured hippocampal astrocytes. Brain Res 588: 154–158.
Uehara Y, Nipper V, McCall AL (1997). Chronic insulin hypoglycemia induces GLUT-3 protein in rat brain neurons. Am J Physiol 272: E716–E719.
Uemura E, Greenlee HW (2001). Amyloid beta-peptide inhibits neuronal glucose uptake by preventing exocytosis. Exp Neurol 170: 270–276.
Urabe T, Hattori N, Nagamatsu S, Sawa H, Mizuno Y (1996). Expression of glucose transporters in rat brain following transient focal ischemic injury. J Neurochem 67: 265–271.
Uryu S, Harada J, Hisamoto M, Oda T (2002). Troglitazone inhibits both post-glutamate neurotoxicity and low-potassium-induced apoptosis in cerebellar granule neurons. Brain Res 924: 229–236.
Vannucci SJ, Maher F, Simpson IA (1997). Glucose transporter proteins in brain: delivery of glucose to neurons and glia. Glia 21: 2–21.
Virgin CE, Ha TP, Packan DR, Tombaugh GC, Yang SH, Horner HC et al (1991). Glucocorticoids inhibit glucose transport and glutamate uptake in hippocampal astrocytes: implications for glucocorticoid neurotoxicity. J Neurochem 4: 1422–1428.
Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA, Vane JR (1996). Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc Natl Acad Sci 96: 7563–7568.
Weinstock M, Shoham S (2004). Rat models of dementia based on reductions in regional glucose metabolism, cerebral blood flow and cytochrome oxidase activity. J Neural Transm 111: 347–366.
Yusim A, Franklin L, Brooke S, Ajilore O, Sapolsky RM (2000). Glucocorticoids exacerbate the deleterious effects of gp120 in hippocampal and cortical explants. J Neurochem 74: 1000–1007.
Zhao X, Ou Z, Grotta JC, Waxham N, Aronowski J (2006). Peroxisome-proliferator-activated receptor-gamma (PPARgamma) activation protects neurons from NMDA excitotoxicity. Brain Res 1073–1074: 460–469.
Acknowledgements
This work was supported by Spanish Ministries of Education & Science (SAF2004-00027) and Fundación Santander/Complutense (PR27/05-14063). FPI (BGB), and FPU (JRC and BGPN).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
García-Bueno, B., Caso, J., Pérez-Nievas, B. et al. Effects of Peroxisome Proliferator-Activated Receptor Gamma Agonists on Brain Glucose and Glutamate Transporters after Stress in Rats. Neuropsychopharmacol 32, 1251–1260 (2007). https://doi.org/10.1038/sj.npp.1301252
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301252
Keywords
This article is cited by
-
Therapeutic modulation of JAK-STAT, mTOR, and PPAR-γ signaling in neurological dysfunctions
Journal of Molecular Medicine (2023)
-
Tetrahydrocannabinol and cannabidiol medicines for chronic pain and mental health conditions
Inflammopharmacology (2022)
-
Role of JAK-STAT and PPAR-Gamma Signalling Modulators in the Prevention of Autism and Neurological Dysfunctions
Molecular Neurobiology (2022)
-
Chronic Mild Stress Alters Kynurenine Pathways Changing the Glutamate Neurotransmission in Frontal Cortex of Rats
Molecular Neurobiology (2019)
-
Thermodynamics in Neurodegenerative Diseases: Interplay Between Canonical WNT/Beta-Catenin Pathway–PPAR Gamma, Energy Metabolism and Circadian Rhythms
NeuroMolecular Medicine (2018)
