
Abstract
Fragile X syndrome is due to an expanded CGG repeat in the 5′ UTR of the FMR1 gene. According to repeat size, we distinguish four allele categories: normal (<40 CGG), intermediate (46–60 CGG), premutated (55–200 CGG) and full mutated (>200 CGG). However, the boundaries among these categories are unclear, making it difficult to classify unstable alleles and to estimate the risk of expansion. We report a family with a proband, carrying a methylated full mutation with an amplification of 1.2 kb. PCR analysis demonstrated two alleles of 29 and 61 CGGs in the mother. Sequencing of the 61 CGG allele showed no AGG interruptions. Both mother's sisters had two alleles of 31 and 44 CGGs, and the daughter of one of these had two alleles of 22 and 44 repeats, demonstrating stable transmission of the 44 CGG allele. The maternal grandfather was deceased, but haplotype reconstruction using markers DXS548 and FRAXAC1 demonstrated that he was carrier of the premutated allele. Furthermore, molecular analysis confirmed the same paternity with a probability of 99.79% for all the three sisters. According to these findings, it is likely that the maternal grandfather carried the 44 CGG allele, showing unstable transmission, given that it expanded first to 61 CGGs in one daughter, and then to full mutation in her child. Although we cannot exclude paternal mosaicism, it is likely that a rare event of progression from an intermediate to a premutated and on to a full mutated allele occurred in this family over two generations.
Similar content being viewed by others

Introduction
Fragile X syndrome (FXS), the most common form of X-linked mental retardation, results from an expansion of the CGG repeat sequence at the 5′ UTR of the FMR1 gene. The sequence is highly polymorphic, allowing the distinction of four classes of alleles, according to size. Normal alleles range in size from 6 to ∼50 CGG repeats, with a mode of 29; intermediate alleles, sometimes referred to as protomutated or ‘grey zone’ alleles, vary in length between ∼45 and ∼60 repeats; premutated alleles range from ∼55 to ∼200 repeats; fully mutated alleles exceed 200 repeats and cytosines in CpG sites become methylated. This latter epigenetic change results in inhibition of gene expression, absence of the FMR1 protein and clinical manifestation of the syndrome. The repeat numbers indicated above, which we have taken from the most recent and authoritative report on the subject,1 are not unquestionable and await further definition.
While the distinction between premutation and full mutation is clear, those between premutated and intermediate alleles and between intermediate and normal alleles are unclear, reflecting our ignorance of the basic mechanisms that permit transition from one class to another. A multistep mutational pathway has been hypothesized by Morton and Macpherson,2 to explain the progression of a stable allele to its expanded full mutation state. These authors have suggested a model with four allelic states: N → S → Z → L. First, the initial mutation occurs on a common-sized repeat allele (N), which acquires limited instability. Next, this allele (S) expands or contracts by one or two repeats, and this unstable state may persist for as many as 90 generations.3 However, by repeated expansions, the S allele eventually reaches the premutation size (Z) and can easily hyperexpand to the full mutation range (L). The different transition rates between each mutational stage result in linkage disequilibrium, explaining the occurrence of a founder effect.4 Such an effect has been demonstrated in several populations mainly by haplotyping fragile X and control chromosomes with microsatellite markers DXS548 and FRAXAC1.5
Chiurazzi et al5 investigated a sample of 137 fragile X males from Italy and found that the haplotype DXS548-2 and FRAXAC1-A (2-A) are most commonly associated with Fragile X syndrome (24%), followed by haplotypes 7-C, 6-D and 3-C, respectively. These four haplotypes make up for 70% of all Italian fragile X chromosomes.
Another source of polymorphism within the CGG repeat sequence is provided by AGG interruptions, usually occurring every 10 CGGs. This sequence variant, together with the size of the alleles and the sex of the carrier, are the key factors influencing the stability of alleles during meiosis.1,6,7 In general, one can state that longer alleles, with fewer AGG interruption, going through female meiosis, tend to be unstable and to progress to a larger size.8 Premutations are highly unstable and entail a risk of expansion to full mutation, when transmitted by a female carrier, which is proportional to their size.1 Recent observations suggest that the loss of AGG interruptions is not the initial event which confers instability. It is more likely that this loss is a late event in the expansion process and possibly the last barrier before hyperexpansion.9 The stability (or instability) of intermediate alleles represents a more complex issue. In general, these alleles tend to be stable and only occasionally undergo small expansions or contractions. However, a few of these ‘must’ have a tendency to expand to pre- and eventually to full mutation, if the prevalence of the FXS is to be maintained stable. This unresolved issue has clear practical implications, if one considers that significant numbers of carriers of intermediate or ‘large’ normal alleles are being identified through population screenings. In order to provide new data that might help in clarifying this still obscure issue, we report on an FXS family in which an intermediate allele of 44 repeats expanded to full mutation over two generations.
Family report
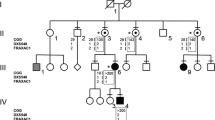
A three-generation-family (Figure 1) was brought to our attention through the propositus (IV-1), an 8-year-old boy, referred because of mental retardation, hyperactivity and physical features consistent with a diagnosis of FXS. The mother had two previous miscarriages. No other cases of mental retardation are reported in the family. The propositus’ mother was 45 and one of her sisters 43 years old. Neither had experienced premature ovarian failure. The molecular analysis of FMR1 was extended from the propositus to other members of the family. All involved individuals gave informed consent to this study, either directly or through their parents.

Pedigree of the family with DXS548 and FRAXAC1 alleles and with the number of FMR1 CGG repeats indicated beneath the symbol of tested individuals. Next to the propositus (IV-1), ‘full’ stands for his full mutation.
Materials and methods
Southern blot analysis
Genomic DNA was isolated from 10 ml of whole blood, using standard procedures. Analysis of the FRAXA expansion was carried out by double digestion of genomic DNA (10 μg) with HindIII and EagI (New England Biolabs). The digested samples were separated on a 0.8% agarose gel with 1 × TAE buffer, blotted on the Hybond-N+ membrane (Amersham-Pharmacia) and hybridized with P32-labeled Ox1.9 probe.10 After overnight hybridization and subsequent washing, the radioactive filter was exposed at −80°C to a Kodak XAR film.
Sizing of CGG repeats
CGG repeat number was estimated by radioactive PCR with primers ‘c’ and ‘f’, according to Fu et al11 The PCR products were separated on 6% polyacrylamide/7 M urea denaturing gel in 1 × TBE, and the radioactive filter was exposed to a Kodak XAR film at −80°C.
Haplotype analysis and paternity test
The microsatellite primers employed for haplotyping were FRAXAC112 and DXS548.13 In all, 30 PCR cycles (94°/1 min–55°/1 min–72°/1 min) were performed in a hot bonnet PCR cycler; the reaction was carried out in 20 μl with 100 ng genomic DNA, Promega Taq Master Mix, 0.1 μl of alpha-32P-dCTP (3000 Ci/ml), 0.5 μ M of each FRAXAC1 and DXS548 primer. PCR products were denatured and loaded on a 6% polyacrylamide/7 M urea denaturing gel in 1 × TBE. Samples were run at 35 W for ∼3 h depending on the size. The gel was dried and exposed at −80°C to a Kodak XAR film. For the paternity test, DNA samples were coamplified using PowerPlex 16 System (Promega), detecting 16 STR loci. Amplified products were analyzed on ABI Prism 310 Genetic Analyzer DNA Sequencer. Plots were interpreted by the probabilistic expert system FINEX, kindly provided by RG Cowell.14
Sequence analysis
Two stages of PCR were used to amplify the two alleles of female carriers prior to sequence analysis of FMR1 CGG repeat. The first stage consisted of an amplification with Pfu polymerase (Stratagene) using primers ‘c’ and ‘f’.11 Cycling conditions were 98°C for 5 min, followed by 35 cycles of 98°C for 1 min, 65°C for 1 min, 72°C for 2 min and 30 s, with a final extension of 75°C for 10 min. The products were separated on 2% agarose gel in 1 × TAE. Once the fragments were well resolved, the band of interest was isolated from the gel using a Concert™ Rapid gel extraction system (Invitrogen). In the second stage, the PCR product was reamplified with the same conditions of the first amplification, but for 23 cycles. The second PCR product was isolated using the previous method. Amplification products were subcloned into TOPO TA Cloning Kit (Invitrogen) and sequenced with the M13 forward and reverse primers of the TOPO TA vector. Every clone was sequenced with ABI Prism Dye terminator (v3.1). The products after sodium-acetate/ethanol precipitation were analyzed on ABI Prism 3100 Genetic Analyzer DNA Sequencer and sequences were analyzed manually.
Results
Molecular analysis of the FMR1 gene by Southern blot showed the propositus to be carrier of a methylated full mutation with an amplification of 1.2 kb. PCR and sequence analysis on the other family members showed that the propositus’ mother was heterozygous, with a 29 and a 61 CGG repeat allele (Figure 1, III-2). Both her sisters (III-3, III-4) were 31/44 heterozygotes, while a normal brother (III-5) carried a 29 CGG repeat allele. One of the sisters had two children, one boy (IV-2) with an allele of 31 repeats and one 22/44 heterozygous girl (IV-3). The maternal grandmother (II-1) was a 29/31 repeats heterozygote. The maternal grandfather and two of his brothers were deceased. One surviving brother (II-5) carried a 23 CGG allele. Sequencing of the 44 CGG allele from subject IV-3 and of the 61 CGG allele of subject III-2 demonstrated absence of AGG interruptions. A paternity test in the three sisters (III-2, III-3, III-4) was concordant, indicating the same paternity with a probability of 99.79%. The propositus is a carrier of 7-C DXS548-FRAXAC1 haplotype, which cosegregates with the carrier status in other family members, as shown in Figure 1.
Discussion
In spite of lack of information on the maternal grandfather (II-2), the interpretation of the family seems unequivocal. Although it is possible that II-2 had a germinal mosaicism for the 61 CGG allele, it appears more likely that he was carrier of an uninterrupted 44 CGG allele, which was stably transmitted to two of his daughters (III-3 and III-4) and then to a granddaughter (IV-3). On the other hand, the first daughter (III-2) received an expanded allele of 61 pure CGGs, a rare and unexpected event, on account of the small size of the original allele. The expansion from 44 to 61 repeats was likely favored by the lack of AGG interruptions, which usually play a stabilizing role.9 In the present case, expansion of the CGG tract cannot result from unequal crossing-over, since paternal meiosis is involved. One might hypothesize an unequal sister-chromatid exchange, either pre- or postzygotic, which may also explain the exceptional case reported by Macpherson et al15 of a 29-repeat allele expanding to 39 repeats, in the presence of two AGG interruptions. However, we think that the 44–61-repeat expansion in our family was likely due to mitotic replication slippage.16,17 By this event, the propositus’ mother (III-2) became a premutation carrier and founder of a new FXS family. The status of the other women (III-3, III-4 and IV-3) in this family remains unclear. Based on evidence from the literature, it is unlikely that they may have affected children due to expansion of their 44 CGG allele to full mutation. However, they are probably at some risk of transmitting a premutation to their descendents, who should be eventually tested either pre- or postnatally.
According to Ashley-Koch et al,6 mutation rates differ significantly between males and females, due to a different selection process in the sperm and egg. These authors found a higher germline mutation rate in males compared to females, and proposed a mitotic model in which CGG repeat instability occurs in many premeiotic divisions, rather than in the meiotic one.6 Since premeiotic divisions in females are limited to prenatal development, whereas they occur in males throughout their fertile life, there are more chances for replication slippage in males than in females. In fact, Sullivan et al7 performed a large population study and observed a higher instability in male transmissions of intermediate alleles (∼40–59 CGGs). To the best of our knowledge, this is the first case reported in the literature documenting the expansion of an intermediate allele with 44 uninterrupted CGGs to a premutation with 61 repeats.
References
Nolin SL, Brown WT, Gliksman A et al: Expansion of the fragile X CGG repeat in females with premutation or intermediate alleles. Am J Hum Genet 2003; 72: 454–464.
Morton NE, Macpherson JN : Population genetics of the Fragile X syndrome: Multiallelic model for the FMR1 locus. Proc Natl Acad Sci USA 1992; 89: 4215–4217.
Chakravarti A : Fragile X founder effect? Nat Genet 1992; 1: 237–238.
Chiurazzi P, Macpherson J, Sherman S, Neri G : Significance of linkage disequilibrium between the fragile X locus and its flanking markers. Am J Med Genet 1996a; 64: 203–208.
Chiurazzi P, Genuardi M, Kozak L et al: Fragile X founder chromosomes in Italy: A few initial events and possible explanation for their heterogeneity. Am J Med Genet 1996b; 64: 209–215.
Ashley-Koch AE, Robinson H, Glicksman AE et al: Examination of factors associated with instability of the FMR1 CGG repeat. Am J Hum Genet 1998; 63: 776–785.
Sullivan AK, Crawford DC, Scott EH et al: Paternally transmitted FMR1 alleles are less stable than maternally transmitted alleles in the common and intermediate size range. Am J Hum Genet 2002; 70: 1532–1544.
Eichler EE, Holden JJA, Popovich BW et al: Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat Genet 1994; 8: 88–94.
Dombrowski C, Levesque S, Morel ML et al: Premutation and intermediate-size FMR1 alleles in 10572 males from the general population: loss of an AGG interruption is a late event in the generation of Fragile X syndrome alleles. Hum Mol Genet 2002; 11: 371–378.
Nakahori Y, Knight SJL, Holland J et al: Molecular heterogeneity of the fragile X syndrome. Nucleic Acids Res 1991; 19: 4355–4359.
Fu YH, Kuhl DPA, Pizzuti A et al: Variation of the CGG repeat et the Fragile X site results in genetic instability: resolution of the Sherman paradox. Cell 1991; 67: 1047–1058.
Richards RI, Holman K, Kozman H et al: Fragile X syndrome: Genetic localization by linkage mapping of two microsatellite repeats FRAXAC1 and FRAXAC2 which immediately flank the fragile site. J Med Genet 1991; 28: 818–823.
Riggins GJ, Sherman SL, Oostra BA et al: Characterization of a highly polymorphic dinucleotide repeat 150 kb proximal to the Fragile X site. Am J Med Genet 1992; 43: 237–243.
Cowell RG : FINEX: A probabilistic expert system for forensic identification. Foren Sci Int 2003; 134: 196–206.
Macpherson JN, Curtis G, Crolla JA et al: Unusual (CGG)n expansion and recombination in a family with fragile X and DiGeorge syndrome. J Med Genet 1995; 32: 236–239.
Mornet E, Chateau C, Hirst MC et al: Analysis of germline variation at the FMR1 CGG repeat shows variation in the normal–premutated borderline range. Hum Mol Genet 1996; 5: 821–825.
Nolin SL, Houck GE, Gargano AD et al: FMR1 CGG-repeat instability in single sperm and lymphocytes of fragile-X premutation males. Am J Hum Genet 1999; 65: 680–688.
Acknowledgements
This work was partly supported by MIUR of Italy and by Fondazione Cariplo.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Terracciano, A., Pomponi, M., Marino, G. et al. Expansion to full mutation of a FMR1 intermediate allele over two generations. Eur J Hum Genet 12, 333–336 (2004). https://doi.org/10.1038/sj.ejhg.5201154
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201154
Keywords
This article is cited by
-
EMQN best practice guidelines for the molecular genetic testing and reporting of fragile X syndrome and other fragile X-associated disorders
European Journal of Human Genetics (2015)
-
A genetic study of the FMR1 gene in a Sardinian multiple sclerosis population
Neurological Sciences (2015)
-
A Dual-Mode Single-Molecule Fluorescence Assay for the Detection of Expanded CGG Repeats in Fragile X Syndrome
Molecular Biotechnology (2013)
-
Couples at risk for spinocerebellar ataxia type 2: the Cuban prenatal diagnosis experience
Journal of Community Genetics (2013)
-
Frühzeitige Ovarialinsuffizienz und FMR1-Mutation
Der Gynäkologe (2012)
