
Abstract
The mechanism of p53-dependent apoptosis is still only partly defined. Using early-passage embryonic fibroblasts (MEF) from wild-type (wt), p53−/− and bax−/− mice, we observe a p53-dependent translocation of Bax to the mitochondria and a release of mitochondrial Cytochrome c during stress-induced apoptosis. These events proceed independent of zVAD-inhibitable caspase activation, are not prevented by dominant negative FADD (DN-FADD), but are negatively regulated by Mdm-2. Bcl-xL expression prevents the release of mitochondrial Cytochrome c and apoptosis, but not Bax translocation. At a single-cell level, enforced expression of p53 is sufficient to induce Bax translocation and Cytochrome c release. Real-time RT-PCR analysis reveals a significant induction of RNA expression of Noxa and Bax in p53+/+, but not in p53−/− MEF. Noxa protein expression becomes detectable prior to Bax translocation, and downregulation of endogenous Noxa by RNA interference protects wt MEF against p53-dependent apoptosis. Hence, in oncogene-expressing MEF p53 induces apoptosis by BH3 protein-dependent caspase activation.
Similar content being viewed by others


Introduction
The activation of oncogenes sensitizes primary cells towards a p53-dependent stress response, which includes cell cycle arrest, cellular senescence and apoptosis. In order to overcome this endogenous defense mechanism, immortalized cells are strongly selected for mutations of the p53 pathway.1,2,3 This observation is supported in vitro and in vivo by genetic mouse models, which demonstrate enhanced tumorigenesis as well as increased resistance towards various stresses or anticancer treatment on the background of a deficiency for p53 and the gene products of the INK4A/ARF locus.4,5,6,7,8,9,10 Additionally, in some experimental systems, endogenously arising murine tumors are selected for mutations of p53 and ARF, or for amplification of the p53 repressor Mdm-2.8 This confirms the functional importance of the p53 tumor suppressor in vivo, which is implicated by its frequent mutation in human cancers.11 Further understanding of the molecular events of these genetically defined pathways will help to devise specific strategies for cancer treatment and prevention.
While the p53-dependent cell cycle checkpoints are well characterized, the actual mechanism whereby p53 activates programmed cell death is still not fully understood.3 Through its activity as a sequence-specific transcription factor, p53 can induce expression of several proapoptotic genes including the death receptors CD95/Fas/APO-1,12,13 KILLER/DR5,14 and the proapoptotic Bcl-2 family members Bax,15 Noxa16 and PUMA.17,18 Additional genes, such as the PIGs,19 PIDD,20 IGF-Binding Protein 3,21 PERP22 or p53AIP1,23 were identified to be transcriptionally upregulated during p53-dependent cell death. However, their roles in the apoptotic pathways, as they are currently understood, remain to be defined.
Changes in the intracellular localization of molecules are crucial to the regulation of apoptosis. The ‘mitochondrial’ apoptotic pathway is activated upon the formation of the Apaf-1-Caspase 9 ‘apoptosome’, which requires release of Cytochrome c from the mitochondrial intermembrane space into the cytoplasma.24 In addition, the proapoptotic molecules Smac/DIABLO and HtrA2/Omi are released from the mitochondria most likely to derepress caspase activation through interaction with IAP proteins.25,26,27,28,29,30 Mitochondrial Cytochrome c release is regulated by the various pro- and antiapoptotic Bcl-2 family members.31 It can either be triggered via the death receptor pathway, involving the cleavage and activation of proapoptotic Bid by Caspase 8,32,33,34,35 or by the stress pathway, which activates proapoptotic Bcl-2 family members through an unknown mechanism. Recently, it has been reported that during death receptor-dependent and -independent apoptosis the proapoptotic Bcl-2 family member Bax undergoes a conformational change, followed by its redistribution from the cytoplasma to the outer mitochondrial membrane, into which it inserts to induce the release of Cytochrome c.36,37,38,39,40 Bim,41 another proapoptotic Bcl-2 family member, is also regulated via its subcellular localization. Upon certain stresses, Bim translocates from the microtubule-associated dynein motor complex to the mitochondria and cellular membranes to promote caspase activation and apoptosis.42
We have previously shown in cancer cell lines that p53 induces apoptosis by a death receptor-independent induction of Cytochrome c release.43 Moreover, primary murine cells lacking Cytochrome c,44 Apaf-145,46,47 or Caspase 9,47,48,49 all essential components of the ‘mitochondrial’ pathway, were protected against stress- or oncogene-induced apoptosis. In the present work, we attempt to further delineate the p53-dependent apoptotic pathway in a genetically defined tumor model. We address the hypothesis that p53 triggers cell death by caspase-independent regulation of the activation and intracellular localization of proapoptotic Bcl-2 family proteins.
Results
Bax is involved in p53-dependent apoptosis and caspase activation of oncogene-expressing murine embryonic fibroblasts
In a cell-free system using extracts from Saos-2 osteosarcoma cells, we showed that the p53-induced release of mitochondrial Cytochrome c requires cytosolic Bax.43 In order to extend this observation in vivo, we obtained MEF deficient in p53 or Bax,50 and their respective wild-type controls, and retrovirally transduced those cells to express the oncogenes E1A, Myc and H-rasG12 V. Following short-term selection, the MEF were treated under various conditions to induce apoptosis. In agreement with previous observations,51 p53-deficient MEF as well as Bax-deficient MEF were protected against apoptosis induced by DNA-damaging agents and UV radiation (Figure 1a). To study the requirement of caspase activation in this system, wt MEF were treated with proapoptotic stimuli in the presence or absence of the broad-spectrum caspase inhibitor zVAD-fmk. Blocking caspase activity effectively prevented the onset of cell death (Figure 1a), confirming its requirement for p53-dependent apoptosis in fibroblasts.52 The extent of death observed in p53−/− or bax−/− MEF at low and intermediate stress intensity roughly equaled the amount of cell death found in wt MEF in the absence of caspase activity (Figure 1a). Thus, caspase activation in oncogene-expressing MEF by low or intermediate doses of DNA damage depended on p53 and Bax. At high doses, however, p53−/− and bax−/− MEF were only partly protected as compared to caspase inhibition by zVAD-fmk (Figure 1a). This argues for p53- and Bax-independent pathways of caspase activation in MEF, which can be triggered by very high doses of DNA-damaging agents. Similar observations have been reported in cancer cell lines.53

Stress-induced apoptosis of oncogene-expressing MEF involves p53 and Bax signaling. (a) Early-passage MEF of the respective genotype expressing E1A and H-rasG12 V were treated with cisplatin (upper), etoposide (middle) or UV radiation (lower panel). In addition, wt MEF were also treated in the presence of the broad-spectrum caspase inhibitor zVAD-fmk, and cell death was assessed by flow cytometry (sub-G1 fraction) 24 h following treatment. Similar results were obtained by annexin-V-PI staining (not shown). Mean values were normalized47 to the rate of spontaneous cell death of untreated early-passage MEF (<10% in transformed p53−/− and bax−/− MEF, and <25% in transformed wt MEF). (b) Early-passage MEF were retrovirally transduced to express Myc and H-rasG12 V, and were plated at low densities in 35 mm dishes. After selection for 1 week, cells were fixed and stained for analysis. A representative result of three independent experiments is shown
The Myc-induced apoptotic response of fibroblasts was found to involve the activation of death receptor pathways as well as mitochondrial Cytochrome c release.54 Furthermore, it was shown to require the formation of the Apaf-1-Caspase 9 ‘apoptosome’, as Myc-transformed fibroblasts from mice deficient in Apaf-1 or Caspase 9 were largely protected against stress-induced, p53-dependent apoptosis.47 Accordingly, the clonogenic growth of early-passage MEF expressing Myc and H-rasG12 V was rescued by p53-deficiency, and was partially rescued by Bax-deficiency. Enumeration revealed that p53−/− MEF formed 25.9 (±4) times more foci, and bax−/− MEF formed 12.1 (±2) times more foci than wt MEF following expression of c-Myc and H-rasG12 V (Figure 1b).
In cancer cell lines or in cell-free reconstituted systems, Bax induces the release of Cytochrome c from the mitochondrial intermembrane space into the cytosol.55,56 This enables the formation of the Apaf-1–Caspase 9 ‘apoptosome’, which activates the effector caspases.31,57 To study these events in oncogene-expression MEF, subcellular fractionation was performed before and after 24 h of treatment with etoposide, and the mitochondria-containing fractions were analyzed by immunoblotting. Following etoposide treatment, a loss of mitochondrial Cytochrome c was observed only in wt MEF, but not in p53−/− or bax−/− MEF (Figure 2a). The release of Cytochrome c corresponded with caspase activation, as indicated by the detection of processed Caspase 3 and the cleavage of the caspase substrate PARP in treated wt MEF (Figure 2a). Time course experiments demonstrated that the onset of p53-dependent apoptosis paralleled the release of mitochondrial Cytochrome c (Figure 2b). Despite low-level apoptosis, no Cytochrome c release was observed following etoposide treatment of bax−/− MEF, whereas at a late time point, some Cytochrome c was lost from the mitochondria of p53−/− MEF (Figure 2b).

Stress-induced apoptosis of oncogene-expressing MEF involves Bax-dependent mitochondrial Cytochrome c release and caspase activation. (a) Oncogene-expressing MEF were treated with 2.5 μM etoposide. Floating and adherent cells were harvested before (−) and 24 h after (+) treatment, the mitochondria-containing fraction was obtained and analyzed by immunoblotting with the indicated primary antibodies. Note the loss of mitochondrial Cytochrome c, the appearance of processed Caspase 3 and the degradation of PARP in treated wt MEF. (b) Oncogene-expressing MEF were treated with 2.5 μM etoposide, and the fraction of apoptotic cells with sub-G1 DNA content (mean±S.D.) was determined by cell cycle analysis and flow cytometry (upper panel). In parallel, the mitochondria-containing fraction was obtained from identically treated MEF, and was analyzed by immunoblotting for the loss of Cytochrome c (C). The blots were reprobed for actin (A) as loading control (lower panel)
Taken together, the p53-dependent apoptotic response of oncogene-expressing early-passage MEF at least in part involves caspase activation through the proapoptotic Bcl-2 family member Bax and mitochondrial Cytochrome c release.
Cytosolic Bax redistributes to mitochondria during p53-dependent apoptosis of oncogene-expressing MEF, independent of Cytochrome c release or caspase activation
The release of mitochondrial Cytochrome c can be directly induced by the proapoptotic Bcl-2 family member Bax.55,56 In vivo, this activity appears to require a conformational change of Bax, which then enables its homo-oligomerization and insertion into the outer mitochondrial membrane.58,59,60 Accordingly, we found an accumulation of Bax in the mitochondria-containing fraction following etoposide treatment of wt MEF, but not of p53−/− or bax−/− MEF (Figure 3). Both, mitochondrial accumulation of Bax and Cytochrome c release, were not prevented by the caspase inhibitor zVAD-fmk or by expression of DN-FADD, which is consistent with a death receptor-independent and caspase-independent engagement of the mitochondrial pathway by p53. This contrasts reports implying the activation of Caspase 8 as an upstream event in p53-dependent apoptosis in different experimental systems.61,62,63

p53-dependent Bax translocation and Cytochrome c release in oncogene-expressing MEF. Early-passage oncogene-expressing MEF of the indicated genotypes and wt MEF expressing DN-FADD, Mdm-2 or Bcl-xL were left untreated (−), or were treated with 2.5 μM etoposide (+) or 2.5 μM etoposide and zVAD-fmk (z). Floating and adherent cells were harvested after 18 h of treatment, and fractionated as described. The cytosolic and mitochondria-containing fractions were analyzed by immunoblotting using the respective primary antibodies. Note the redistribution of Bax from the cytosolic to the mitochondrial fraction in wt, DN-FADD and Bcl-xL-expressing MEF, and the loss of mitochondrial Cytochrome c in wt and DN-FADD-expressing MEF
p53 deficiency or expression of Mdm-2, which destabilizes p53 by targeting for proteasomic degradation,64 prevented mitochondrial accumulation of Bax and Cytochrome c release. Again, no Cytochrome c release was observed in bax−/− MEF under the present experimental conditions (Figure 3). In contrast, expression of Bcl-xL, which effectively prevents the release of mitochondrial Cytochrome c (Figure 3) and the induction of apoptosis (not shown), did not influence the mitochondrial translocation of Bax. Similar to a recent study using human fibroblasts,65 neither mitochondrial Cytochome c release, nor Bax translocation were observed upon etoposide treatment of nontransformed MEF (data not shown).
These results are consistent with a model of stress-induced apoptosis, in which activated p53 signals to Bax (or other proapoptotic Bcl-2 proteins) to engage the ‘mitochondrial’ death pathway independent of caspase activation or death receptor signals. Activation of the caspase cascade seems to occur downstream of mitochondria, most likely via the formation of the Apaf-1–Caspase 9 ‘apoptosome’.47
p53 expression is sufficient to induce caspase-independent translocation of Bax from the cytosol to mitochondria, and release of Cytochrome c from the mitochondria into the cytosol
To study the changes of intracellular localization of Cytochrome c and Bax during p53-dependent apoptosis at a single-cell level, we devised HeLa cells, and adenovirus-mediated p53 gene transfer.43 Previously, it has been reported that adenovirus-mediated expression of p53 can induce apoptosis in HeLa cells independent of Bax translocation and Cytochrome c release.66 Using a similar adenoviral p53 vector, we confirmed that p53 expression can lead to apoptosis of HeLa cells (Figure 4). However, by means of time-lapse confocal microscopy,67 we found that the release of Green-fluorescent protein (GFP)-tagged Cytochrome c from the mitochondria into the cytosol always preceded apoptotic cell death of HeLa cells induced by the expression of p53 (Figure 5a). The p53-induced release of Cytochrome c in HeLa cells occurred independent of caspase activity, as treatment with the broad-spectrum caspase inhibitor zVAD-fmk directly after adenoviral transduction could not prevent the release of GFP-Cytochrome c from mitochondria to the cytosol (Figure 5a).

Adenovirus-mediated expression of p53 induces apoptosis in HeLa cells. HeLa cells were adenovirally transduced (5 × 109 viral particles/ml for 1 h, as described43) to express high levels of p53 (Ad.p53) or control vector (Ad). Apoptosis was measured at the indicated time points following transduction by staining with annexin-V-FITC and PI, and dual-color flow cytometry. The mean percentage (±S.D.) of annexin-V-negative/PI-negative viable cells is given

p53 expression, induces caspase-independent redistribution of Bax and release of mitochondrial Cytochrome c. (a) A time lapse of HeLa cells undergoing apoptosis following adenovirus-mediated p53 expression. GFP-Cytochrome c moves from a mitochondrial staining pattern to a diffuse cytoplasmic distribution in the presence of the broad-spectrum caspase inhibitor zVAD-fmk. The arrow indicates a Cytochrome c-releasing cell; the number at the bottom right of each frame represents the time after transduction. Images were taken with a 40 × objective and a zoom of 2. (b) A time lapse of GFP-Bax localization following p53 expression. HeLa cells with a mostly diffuse cytoplasmic, but some perinuclear GFP-Bax localization change to a perinuclear pattern in the presence of zVAD-fmk. The arrow indicates a cell with Bax translocation to the mitochondria; the number at the bottom of each frame represents the time after transduction. Images were taken with a 40 × objective and zoomed 1.75 times. (c) Perinuclear GFP-Bax colocalizes with a mitochondria stain. Left image (green) is GFP-Bax following translocation. Middle image (red) is the mitochondrial marker TMRE (40 μM). The result of a merge of the left and middle images is on the right (40 × objective, zoom 3). The colocalization of GFP-Cytochrome c with mitochondria in nonapoptotic cells was established previously.67
To study whether p53 expression is sufficient to induce the mitochondrial translocation of Bax, we stably expressed GFP-tagged Bax68 in HeLa cells. When apoptosis was induced by adenoviral expression of p53, GFP-Bax was redistributed from a diffuse pattern to a predominantly perinuclear, punctate pattern (Figure 5b). Following redistribution, the GFP-Bax signal colocalized with the signal obtained by the mitochondrial dye TMRE (Figure 5c), suggesting a translocation of GFP-Bax from the cytosol to mitochondria in response to p53 expression. Like p53-induced Cytochrome c release, Bax translocation also occurred in the presence of the caspase inhibitor zVAD-fmk, ordering this event upstream of zVAD-inhibitable caspase activation in p53-induced apoptosis.
Similar results were obtained by immunofluorescence detection of endogenous Bax in oncogene-expressing MEF treated with various apoptotic stimuli. A conformational change of Bax, which precedes its mitochondrial translocation, and which can be detected by an antiserum specific against the N-terminus of the molecule,37,40 was induced by DNA-damaging agents in the absence of caspase activation (data not shown).
Induction of the BH3-protein Noxa is essential for p53-dependent apoptosis of oncogene-expressing fibroblasts
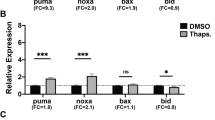
p53 is thought to exert most of its functions through its activity as a sequence-specific transcriptional activator.1 In order to assess the impact of p53 on the RNA expression of proapoptotic molecules, we isolated and reverse-transcribed RNA from oncogene-expressing wt and p53−/− MEF following treatment with etoposide. By quantitative real-time PCR analysis, we found a dramatic induction of Bax and the BH3-domain containing Noxa16 in wt MEF. RNA expression peaked at 7 h post-treatment and decreased later on (Figure 6a, and not shown). In addition, the BH3-protein PUMA,17,18 the zinc finger protein Peg3,69 and the p53 target gene p21/WAF170 were induced in a p53-dependent manner (Figure 6a). In contrast, the expression of proapoptotic Bak, which acts at the same level as Bax during stress-induced apoptosis in MEF,71,72 remained unchanged following etoposide treatment (Figure 6a). Time-course experiments revealed that the RNA induction of Noxa and Bax occur as early as 3 h after etoposide treatment, which is prior to Bax translocation and mitochondrial Cytochrome c release, that become detectable after 8 h of treatment (Figure 6b). At the protein level, Noxa expression became detectable after 4 h treatment with etoposide, and thus earlier than the redistribution of Bax and Cytochrome c can be observed (Figure 6b).

Induction of Noxa is necessary for p53-dependent apoptosis of oncogene-expressing MEF following DNA damage. (a) Oncogene-expressing wt (closed squares) and p53−/− (open squares) MEF were treated with 2.5 μM etoposide, and total RNA was isolated at the respective time points. Following reverse transcription, the expression of BAX, BAK, PUMA, NOXA, PEG3, p21/WAF1 (a), BCL-2, ACTIN and 18S RNA (not shown) was quantitated by real-time PCR as described. Mean values of triplicates of a representative experiment are shown. (b) (Upper panel) Timing and relative induction of genes implicated in mitochondrial Cytochrome c release in oncogene-expressing wt MEF treated with 2.5 μM etoposide. (Lower panel) Timing of protein expression and redistribution of Cytochrome c and Bax in oncogene-expressing wt MEF following treatment with 2.5 μM etoposide. Note the decrease of cytosolic Bax and the increase of cytosolic Cytochrome c following 8 h of treatment. (c) (Upper panel) MEF-expressing RNA oligonucleotides interfering with Noxa (open diamonds, light bars) or irrelevant control genes (closed squares, open triangles, dark bars) were treated with etoposide (VP16), UV radiation (UV), staurosporine (STS) or actinomycin D (actD). Apoptosis was measured by flow cytometry (sub-G1 fraction) 24 h following treatment. (Lower panel) Expression of endogenous p53, Bax and Noxa in parental wt MEF (mock), and MEF expressing RNA oligonucleotides interfering with Noxa (noxa) and two control oligonucleotides (C1, C2)
To assess whether the induction of Noxa is essential for p53-dependent apoptosis of oncogene-expressing MEF, we devised RNA interference73 to knock down endogenous Noxa RNA expression in our experimental system. Stable expression of Noxa-specific interfering RNA oligonucleotides74,75 protected oncogene-expressing wt MEF against p53-dependent apoptosis induced by etoposide, UV radiation, staurosporine and actinomycin D (Figure 6c). Hence, the induction of the BH3-only protein Noxa, which indirectly acts on mitochondria via Bax or Bak,76 is essential for p53-dependent apoptosis in the present experimental system.
Discussion
Depending on the cellular context, the p53 tumor suppressor protein engages several stress response pathways including cell cycle arrest, cellular senescence and apoptosis.1,2 The molecular mechanism whereby p53 signals to the apoptotic machinery, however, is still not completely understood.3 Using genetically defined murine fibroblasts, which were sensitized to apoptosis by the expression of oncogenes, we identified conditions, under which stress-induced cell death is dependent on p53, the proapoptotic Bcl-2 family protein Bax and caspase activation. In this system, apoptotic p53 signaling triggers the ‘mitochondrial’ pathway, as indicated by the release of Cytochrome c into the cytoplasm in the absence of zVAD-inhibitable caspase activation. Mitochondrial Cytochrome c release is mediated by proapoptotic Bcl-2 family proteins, such as Bax or Bak,57 and was prevented in bax−/− MEF under our experimental conditions.
In cancer cell lines, proapoptotic stimuli result in a redistribution of the Bcl-2 family proteins Bax36,37,38,39,40 and Bim42 from the cytoplasm or microtubules to the mitochondria. This relocalization appears to be a prerequisite for caspase activation via mitochondrial Cytochrome c release. In the present study, we show that p53 activation leads to a translocation of Bax from the cytoplasm to the mitochondria. Bax translocation was inhibitable by Mdm-2, but not by Bcl-xL or by the broad-spectrum caspase inhibitor zVAD-fmk. A similar observation was recently reported using Myc-transformed fibroblasts expressing a temperature-sensitive p53 mutant.77 In this study, p53-dependent transcriptional activation of the zinc finger gene Peg3/Pw1 was implicated to mediate activation and mitochondrial translocation of Bax. Alternatively, p53-regulated BH3-only proteins,78 such as PUMA and Noxa, might act as activators of the proapoptotic ‘multidomain’ Bcl-2 proteins Bax and Bak.76,79 By quantitative real-time RT-PCR analysis, we found a p53-dependent induction of Bax and Noxa RNA following etoposide treatment of oncogene-expressing MEF. RNA expression of PUMA and Peg3/Pw1 was also significantly induced in wt MEF, however, to a much lesser extent than the former genes. Comparing the timing of gene induction with the order of events at the protein level, the expression of the BH3-only protein Noxa preceded Bax translocation and Cytochrome c release, as detected by cell fractionation and immunoblotting. Using an RNAi approach,74 we demonstrated that endogenous Noxa is necessary for p53-dependent apoptosis induced by DNA damage in oncogene-expressing fibroblasts. These results are consistent with a model, in which p53 signals apoptosis through the BH3-protein Noxa, which then activates the ‘mitochondrial’ pathway of caspase activation through Bax in oncogene-expressing MEF, or additional proapoptotic ‘multidomain’ Bcl-2 proteins in other experimental systems.72,79
Our results using quantitative RT-PCR analysis are suggestive of a mechanism involving p53-dependent transcriptional activation of the Noxa gene as the most upstream event in this cascade. However, alternative mechanisms of activation of BH3-proteins have been described.32,42 Moreover, stress-induced p53-dependent apoptosis can proceed independent of transcription or translation in some experimental systems.80,81,82 In addition, Bax-independent activation of stress-induced apoptosis by certain cytotoxic agents has been reported.53 Thus, it is possible that p53 can activate several proapoptotic Bcl-2 family members through alternative or parallel pathways leading to mitochondrial Cytochrome c release and apoptosis. This complexity, however, may explain the discrepant results obtained in various experimental systems studying the molecular events of p53-dependent apoptosis.
Materials and Methods
Cell culture
Fibroblasts from 2-week-old p53−/− murine embryos and their wild-type littermates were prepared according to standard techniques. Early-passage MEF from bax−/− mice were generously provided by Dr. SJ Korsmeyer.50 Cells were expanded in DMEM containing 15% fetal bovine serum (FBS), L-glutamine, antibiotics and β-mercaptoethanol, and were transduced at passage 4 using high-titer retroviral vectors as previously described.51,83 The HeLa cervical carcinoma cell line was obtained from ATCC (Manassas, VA, USA), and was maintained in DMEM containing 10% FBS, L-glutamine and antibiotics.
Vectors, antibodies and reagents
Retroviral vectors expressing bcl-xL and DN-FADD were generated by subcloning the respective cDNA into pBabePuro or pBabeHygro. The mdm-2 cDNA was subcloned into pMxIG (a gift from Dr. T Kitamura). Retroviral vectors expressing E1A, H-rasG12 V and Myc were kindly provided by Drs. SW Lowe and GJ Hannon. Adenoviral p53 expression vectors43 and a plasmid expressing a GFP-Cytochrome c fusion protein67 were used as previously described. The pEGFP-bax expression plasmid was a gift from Dr. RJ Youle.68
Detection of protein expression was performed by standard immunoblotting techniques using primary antibodies against p53 (PAb 122 and PAb 240, Pharmingen), Bcl-x (rabbit polyclonal antiserum, Pharmingen), Bax and Bak (rabbit polyclonal IgG, Upstate Biotechnology), Noxa (goat polyclonal antiserum, Santa Cruz), Cytochrome c (clone 7H8.2C12, Pharmingen), PARP (N20, goat polyclonal antiserum, Santa Cruz), caspase 3 (rabbit polyclonal antiserum, Pharmingen) and Actin (clone C4, ICN Pharmaceuticals).
Anticancer drugs were purchased from Sigma, the broad-spectrum caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (zVAD-fmk) from Enzyme System Products, and the mitochondrial stain tetramethyl-Rhodamine-ethylester (TMRE) was obtained from Molecular Probes.
Cell death assays
Cell death was assessed by dual color flow cytometry after staining with annexin-V-FITC (Clontech) and propidium iodide (PI), and by flow cytometry and cell cycle analysis following permeabilization and staining with PI.56
Cell fractionation
Following treatment, adherent and floating cells were harvested and washed with PBS. Cell pellets were dissolved in 2 volumes of mitochondria buffer43 supplemented with 100 μg/ml digitonin (Sigma), and were incubated on ice for 10 min or until <10% of cells excluded trypan blue. Alternatively, cells were broken using a Dounce homogenizer, as described previously.43 Cytosolic supernatants and mitochondria-containing pellets were obtained by centrifugation at 13 000 × g. The pellets were resuspended in lysis buffer, freeze–thawed, vortexed and again centrifuged at 13 000 × g. The resulting supernatants were normalized for protein concentration, boiled in Laemmli buffer, and analyzed by SDS-PAGE and immunoblotting.
Clonogenicity assays
Early-passage MEF were retrovirally transduced to express Myc and H-rasG12 V. The transduction efficacy consistently ranged from 80 to 90% of cells in parallel experiments using a GFP-expressing vector. Cells were plated at 104/well in 35 mm dishes, and were cultured for 7 days in selection media. After washing with PBS, cells were fixed with 70% ethanol for 1 h, and were stained with methylene blue for enumeration.
Fluorescence microscopy
Cells were plated overnight in cell culture dishes containing sterilized coverslips. The next day transductions were performed as described.43 After a 24 h incubation in the presence or absence of the caspase inhibitor zVAD-fmk, the culture dishes were placed in a heating chamber and images were taken in 10 or 15 min intervals using an MRC 1024ES laser scanning confocal microscope (BioRad) as described previously.67 The 488 nm line was attenuated 91 and 94% to image the GFP-Cytochrome c and the GFP-Bax fusion proteins, respectively.
For the detection of the activated conformation of endogenous Bax, oncogene-expressing MEF growing in LabTek chamber slides were treated with various apoptotic stimuli. After 12–18 h, cells were fixed with 4% paraformaldehyde in PBS, and were washed 6 times with blocking buffer (4% BSA, 0.05% saponin in PBS). The slides were incubated overnight at 4°C with a rabbit polyclonal antiserum against the N-terminus of Bax (BAX-NT, Upstate Biotechnology) and/or a monoclonal anti-Cytochrome c antibody (Pharmingen). Texas Red-conjugated anti-rabbit Ig and FITC-conjugated anti-mouse Ig (Santa Cruz) were used as secondary antibodies, and nuclei were counterstained with Hoechst 33342 (Sigma). Images were taken on an Olympus IX-70 inverted microscope at 550, 488 and 390 nm excitation frequency using a digital imaging system, and BAX-NT-positive cells were enumerated.
Reverse transcription and quantitative real-time PCR analysis
Total RNA (tRNA) was isolated from wt and p53−/− MEF by means of the QIAgen RNeasy kit following the manufacturer's instructions. Per time point 2 μg of tRNA was transcribed into cDNA using the Omniscript reverse transcriptase (QIAgen). Real-Time PCR™ was performed using AmpliTaq Gold™ polymerase in a PE Biosystems 5700 Thermocycler using the SyBr Green™ detection protocol as per the manufacturer's instructions. Briefly, 12 ng of total cDNA, 50 nM of each primer and 1X SyBr Green mix were used in a total volume of 25 μl. The primer sequences were murine BCL2 (sense 5′-ACT TCG CAG AGA TGT CCA GTC A-3′ and antisense 5′-TGG CAA AGC GTC CCC TC-3′), murine BAK (sense 5′-GTC ACA AAG TCG CGA GGT TTC-3′ and antisense 5′-ATG GTG GTG GAT GCC GC-3′), murine NOXA (sense 5′-AGG AAG GAA GTT CCG CCG-3′ and antisense 5′-AGC GTT TCT CTC ATC ACA TCA CA-3′), murine BAX (sense 5′-GGA GCA GCT TGG GAG CG-3′ and antisense 5′-AAA AGG CCC CTG TCT TCA TGA-3′), murine PUMA (sense 5′-GCC TCC TTT CTC ACC GAG C-3′ and antisense 5′-GGC CGC AAT CTT TGC A-3′), murine p21/WAF1 (sense 5′-CCG TTG TCT CTT CGG TCC C-3′ and antisense 5′-CAT GAG CGC ATC GCA ATC-3′), murine Peg3 (sense 5′-GCC TCC TTT CTC ACC GAG C-3′ and antisense 5′-TGG CCG CAA TCT TTG CA-3′), 18 S RNA (sense 5′-ATG GTA GTC GCC GTG CCT AC-3′ and antisense 5′-CCG GAA TCG AAC CCTGAT T-3′), and murine beta-actin (sense 5′-TTCGTTGCCGGTCCACA-3′ and antisense 5′-ACCAGCGCAGCGATATCG-3′). All samples were read in triplicate, and values were normalized for baseline expression and for expression of β-actin RNA.
RNA interference
Hairpin oligonucleotides were designed using an Internet tool (oligoengine.com), and were ligated into the pSUPER.retro vector.75 The murine Noxa oligos were NOXA216_S 5′-GAT CCC GGA CGA GTG TGC TCA ACT CTT CAA GAG AGA GTT GAG CAC ACT CGT CCT TTT TGG AAA-3′ and NOXA216_AS 5′-AGC TTT TCC AAA AAG GAC GAG TGT GCT CAA CTC TCT CTT GAA GAG TTC AGC ACA CTC GTC CGG-3′. Retroviral vectors were generated and oncogene-expressing (hygromycin-selected) wt MEF were transduced as described above. Following selection with puromycin, MEF populations expressing oligonucleotides interfering with Noxa or control oligonucleotides were analyzed for protein expression by immunoblotting as described above.
Abbreviations
- DMEM:
-
Dulbecco's modified essential medium
- FADD:
-
Fas-associated death domain
- MEF:
-
murine embryonic fibroblasts
- PARP:
-
poly(ADP-ribose) polymerase
- PCR:
-
polymerase chain reaction
- RNAi:
-
RNA interference
- TMRE:
-
tetramethyl-rhodamine-ethylester
- zVAD-fmk:
-
benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone
References
Vogelstein B, Lane D and Levine AJ (2000) Surfing the p53 network. Nature 408: 307–310
Sherr CJ and DePinho RA (2000) Cellular senescence: mitotic clock or culture shock. Cell 102: 407–410
Vousden KH (2000) p53: death star. Cell 103: 691–694
Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery Jr. CA, Butel JS and Bradley A (1992) Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 356: 215–221
Lowe SW, Ruley HE, Jacks T and Housman DE (1993) p53- dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 74: 957–967
Serrano M, Lee H-W, Chin L, Cordon-Cardo C, Beach D and DePinho RA (1996) Role of the INK4a Locus in tumor suppression and cell mortality. Cell 85: 27–37
Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing HR, Ashmun RA, Grosveld G and Sherr CJ (1997) Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell 91: 649–659
Eischen CM, Weber JD, Roussel MF, Sherr CJ and Cleveland HL (1999) Disruption of the ARF–Mdm2–p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev. 13: 2658–2669
Chin L, Tam L, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, O'Hagan R, Pantginis J, Zhou H, Horner II JW, Cordon-Cardo C, Yancopoulos GD and DePinho RA (1999) Essential role for oncogenic Ras in tumor maintenance. Nature 400: 468–472
Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM and Lowe SW (2002) A senescence program controlled by p53 and p16 INK4a contributes to the outcome of cancer therapy. Cell 109: 335–346
Hollstein M, Rice K, Greenblatt MS, Soussi T, Fuchs R, Sorlie T, Hovig E, Smith Sorensen B, Montesano R and Harris CC (1994) Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 22: 3551–3555
Owen-Schaub LB, Zhang W, Cusack JC, Angelo LS, Santee SM, Fujiwara T, Roth JA, Deisseroth AB, Zhang WW, Kruzel E and Radinsky R (1995) Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol. Cell. Biol. 15: 3032–3040
Müller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M and Krammer PH (1998) p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs. J. Exp. Med. 188: 2033–2045
Wu GS, Burns TF, McDonald III ER, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz SD, Wu G and El-Deiry WS (1997) KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 17: 141–143
Miyashita T and Reed HC (1995) Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 80: 293–299
Oda E, Ohki R, Murasawa H, Nemoto J, Shibue T, Yamashita R, Tokino T, Taniguchi T and Tanaka N (2000) Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288: 1053–1058
Yu J, Zhang L, Hwang PM, Kinzler KW and Vogelstein B (2001) PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 7: 673–682
Nakano K and Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7: 683–694
Polyak K, Xia Y, Zweier JL, Kinzler KW and Vogelstein B (1997) A model for p53 induced apoptosis. Nature 389: 300–305
Lin Y, Ma W and Benchimol S (2000) Pidd, a new death-domain containing protein, is induced by p53 and promotes apoptosis. Nat. Genet. 26: 122–127
Buckbinder L, Talbott R, Velasco-Miguel S, Takenaka I, Faha B, Seizinger BR and Kley N (1995) Induction of the growth inhibitor IGF-binding protein 3 by p53. Nature 377: 646–649
Attardi LD, Reczek EE, Cosmas C, Demicco EG, McCurrach ME, Lowe SW and Jacks T (2000) PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev. 14: 704–718
Oda K, Arkawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y and Taya Y (2000) p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell 102: 849–862
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES and Wang X (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates and apoptotic protease cascade. Cell 91: 479–489
Du C, Fang M, Li Y, Li L and Wang X (2000) Smac, a mitochondrial protein that promotes Cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 102: 33–42
Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly KM, Reid GE, Moritz RL, Simpson RJ and Vaux DL (2000) Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 102: 43–53
Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K and Takahashi R (2001) A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol. Cell 8: 613–621
Hedge R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti I, DuBois G, Lazebnik YA, Zervos AS, Fernandes-Alnemri T and Alnemri ES (2002) Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts IAP-caspase interaction. J. Biol. Chem. 277: 432–438
Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, Savopoulos J, Gray CW, Creasy CL, Dingwall C and Downward J (2002) The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a Reaper-like motif. J. Biol. Chem. 277: 439–444
Verhagen AM, Silke J, Ekert PG, Pakusch M, Kaufmann H, Connolly LM, Day CL, Tikoo A, Burke R, Wrobel C, Moritz RL, Simpson RJ and Vaux DL (2002) HtrA2 promotes cell death through its serine protease activity and its ability to antagonise inhibitor of apoptosis proteins. J. Biol. Chem. 277: 445–454
Green DR and Reed JC (1998) Mitochondria and apoptosis. Science 281: 1309–1312
Luo X, Budihardjo I, Zou H, Slaughter C and Wang X (1998) Bid, a Bcl-2 interacting protein, mediates Cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94: 481–490
Li H, Zhou H, Xu C-J and Yuan J (1988) Cleavage BID by Caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94: 491–501
Gross A, Yin X-M, Wang K, Wei MC, Jockel J, Milliman C, Erdjument-Bromage H, Tempst P and Korsmeyer SJ (1999) Caspase cleaved BID targets mitochondria and is required for Cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J. Biol. Chem. 274: 1156–1163
Yin X-M, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B, Roth KA and Korsmeyer SJ (1999) Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 400: 886–891
Hsu Y-T, Wolter KG and Youle RJ (1997) Cytosol-to-membrane redistribution of Bax and Bcl-xL during apoptosis. Proc. Natl. Acad. Sci. USA 94: 3668–3672
Deshager S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B and Martinou J-C (1999) Bid-induced conformational change of Bax is responsible for mitochondrial Cytochrome c release during apoptosis. J. Cell Biol. 144: 891–901
Nomura M, Shimizu S, Ito T, Narita M, Matsuda H and Tsujimoto Y (1999) Apoptotic cytosol facilitates Bax translocation to mitochondria that involves cytosolic factor regulated by Bcl-2. Cancer Res. 59: 5542–5548
Putcha GV, Deshmukh M and Johnson Jr. EM (2000) Inhibition of apoptotic signaling cascades causes loss of trophic factor dependence during neuronal maturation. J. Cell Biol. 149: 1011–1017
Perez D and White E (2000) TNF-a signals apoptosis through a bid-dependent conformational change in Bax that is inhibited by E1B 19 K. Mol. Cell 6: 53–63
O'Connor L, Strasser A, O'Reilly LA, Hausmann G, Adams JM, Cory S and Huang DCS (1998) Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 17: 384–395
Puthalakath H, Huang DCS, O'Reilly LA, King SM and Strasser A (1999) The proapoptotic activity of the Bcl-2 family member bim is regulated by interaction with the dynein motor complex. Mol. Cell 3: 287–296
Schuler M, Bossy-Wetzel E, Goldstein JC, Fitzgerald P and Green DR (2000) p53 induces apoptosis by caspase activation through mitochondrial Cytochrome c release. J. Biol. Chem. 275: 7337–7342
Li K, Li Y, Shelton JM, Richardson JA, Spencer E, Chen ZJ, Wang X and Williams RS (2000) Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell 101: 389–399
Yoshida H, Kong Y-Y, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM and Mak TW (1998) Apaf-1 is required for mitochondrial pathways of apoptosis and brain development. Cell 94: 739–750
Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA and Gruss P (1988) Apaf-1 (CED-4 Homolog) regulates programmed cell death in mammalian development. Cell 94: 727–737
Soengas MS, Alarcón RM, Yoshida H, Giaccia AJ, Hakem R, Mak TW and Lowe SW (1999) Apaf-1 and Caspase-9 in p53-dependent apoptosis and tumor inhibition. Science 284: 156–159
Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de al Pompa JL, Kagi D, Khoo W, Potter J, Yoshoda R, Kaufman SA, Lowe SW and Mak TW (1998) Differential requirement for Caspase 9 in apoptotic pathways in vivo. Cell 94: 339–352
Kuida K, Haydar TF, Kuan C-Y, Gu Y, Taya C, Karasuyama H, Su MSS, Rakic P and Flavell RA (1998) Reduced apoptosis and Cytochrome c-mediated caspase activation in mice lacking Caspase 9. Cell 94: 325–337
Knudson CM, Tung KSK, Tourtellotte WG, Brown GAJ and Korsmeyer SJ (1995) Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science 270: 96–99
McCurrach ME, Connor TMF, Knudson CM, Korsmeyer SJ and Lowe SW (1997) Bax-deficiency promotes drug resistance and oncogenic transformation by attenuating p53-dependent apoptosis. Proc. Natl. Acad. Sci. USA 94: 2345–2349
Sabbatini P, Han J, Chiou SK, Nicholson DW and White E (1997) Interleukin 1b converting enzyme-like proteases are essential for p53-mediated transcriptionally dependent apoptosis. Cell Growth Differ. 8: 643–653
Zhang L, Yu J, Park BH, Kinzler KW and Vogelstein B (2000) Role of BAX in the apoptotic response to anticancer agents. Science 290: 989–992
Juin P, Hueber A-O, Littlewood T and Evan G (1999) c-Myc-induced sensitization to apoptosis is mediated through cytochrome c release. Genes Dev. 13: 1367–1381
Jürgensmeier JM, Xie A, Deveraux Q, Ellerby L, Bredesen D and Reed JC (1998) Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. USA 95: 4997–5002
Finucane DM, Bossy-Wetzel E, Waterhouse NJ, Cotter TG and Green DR (1999) Bax-induced caspase activation and apoptosis via Cytochrome c release from mitochondria is inhibitable by Bcl-xL. J. Biol. Chem. 274: 2225–2233
Green DR (1998) Apoptotic pathways: roads to ruin. Cell 94: 695–698
Nechushtan A, Smith CL, Hsu Y-T and Youle RJ (1999) Conformation of the Bax C-terminus regulates subcellular location and cell death. EMBO J. 18: 2330–2341
Eskes R, Desagher S, Antonsson B and Martinou J-C (2000) Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol. Cell. Biol. 20: 929–935
Suzuki M, Youle RJ and Tjandra N (2000) Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103: 645–654
Ding H-F, Lin Y-L, McGill G, Juo P, Zhu H, Blenis J, Yuan J and Fisher DE (2000) Essential role for Caspase-8 in transcription-independent apoptosis triggered by p53. J. Biol. Chem. 275: 38905–38911
Kovar H, Jug G, Printz D, Bartl S, Schmid G and Werierska-Gadek J (2000) Characterization of distinct consecutive phases in non-genotoxic p53-induced apoptosis of Ewing tumor cells and the rate-limiting role of caspase 8. Oncogene 19: 4096–4107
Feng Gao C, Ren S, Zhang L, Nakajima T, Ichinose S, Hara T, Koike K and Tsuchida N (2001) Caspase-dependent cytosolic release of Cytochrome c and membrane translocation of Bax in p53-induced apoptosis. Exp. Cell Res. 265: 145–151
Oren M (1999) Regulation of the p53 tumor suppressor protein. J. Biol. Chem. 274: 36031–36034
Duelli DM and Lazebnik YA (2000) Primary cells suppress oncogene-dependent apoptosis. Nat. Cell Biol. 2: 859–862
Li P-F, Dietz R and von Harsdorf R (1999) p53 regulates mitochondrial membrane potential through reactive oxygen species and induces cytochrome c-independent apoptosis blocked by Bcl-2. EMBO J. 18: 6027–6036
Goldstein JC, Waterhouse NJ, Juin P, Evan GI and Green DR (2000) The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat. Cell Biol. 2: 156–162
Wolter KG, Hsu Y-T, Smith CL, Nechushtan A, Xi XG and Youle RJ (1997) Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 139: 1281–1292
Relaix F, Wei X, Li W, Pan J, Lin Y, Bowtell DD, Sassoon DA and Wu X (2000) Pw1/Peg3 is a potential cell death mediator and cooperates with Siah 1a in p53-mediated apoptosis. Proc. Natl. Acad. Sci. USA 97: 2105–2110
El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler K and Vogelstein B (1993) WAF1, a potential mediator of p53 tumor suppression. Cell 75: 817–825
Lindsten T, Ross AJ, King A, Zong W-X, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR and Thompson CB (2000) The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol. Cell 6: 1389–1399
Wei MC, Zong W-X, Cheng EHY, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB and Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730
Paddison PJ and Hannon GJ (2002) RNA interference: the new somatic cell genetics. Cancer Cell 2: 17–23
Brummelkamp TR, Bernards R and Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296: 550–553
Brummelkamp TR, Bernards R and Agami R (2002) Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell 2: 243–247
Cheng EHYA, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T and Korsmeyer SJ (2001) BCL-2, BCL-XL sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol. Cell 8: 705–711
Deng Y and Wu X (2000) Peg3/Pw1 promotes p53-mediated apoptosis by inducing Bax translocation from cytosol to mitochondria. Proc. Natl. Acad. Sci. USA 97: 12050–12055
Huang DCS and Strasser A (2001) BH3-only proteins – essential initiators of apoptotic cell death. Cell 103: 839–842
Zong W-X, Lindsten T, Ross AJ, MacGregor GR and Thompson CB (2001) BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev. 15: 1481–1486
Caelles C, Heimberg A and Karin M (1994) p53-dependent apoptosis in the absence of transcriptional activation of p53-target genes. Nature 370: 220–223
Haupt Y, Rowan S, Shaulian E, Vousden KH and Oren M (1995) Induction of apoptosis in Hela cells by transactivation-deficient p53. Genes Dev. 9: 2170–2183
Chen X, Ko LJ, Jayaraman L and Prives C (1996) p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev. 10: 2438–2451
Serrano M, Lin AW, McCurrach ME, Beach D and Lowe SW (1997) Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602
Acknowledgements
We are indebted to Dr. SJ Korsmeyer for providing cells from Bax-deficient mice, to Dr. T Lin for animal care and to P Fitzgerald for help with cloning. Drs. SW Lowe, GJ Hannon, T Kitamura, M Oren, S Hedrick, DC Maneval, RJ Youle and R Agami are thanked for providing expression vectors.
This work was supported by National Institutes of Health grants GM52735, CA69381, and AI40646 awarded to DRG, and by the Max-Eder-Programm of the Deutsche Krebshilfe to MS (70-2952).
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Melino
Rights and permissions
About this article
Cite this article
Schuler, M., Maurer, U., Goldstein, J. et al. p53 triggers apoptosis in oncogene-expressing fibroblasts by the induction of Noxa and mitochondrial Bax translocation. Cell Death Differ 10, 451–460 (2003). https://doi.org/10.1038/sj.cdd.4401180
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401180
