
Abstract
Since 2004, the clinical impact of monoclonal antibodies (mAbs) targeting the epidermal growth factor receptor (EGFR) on patients with metastatic colorectal cancer (MCRC) has been clearly established. The combination of these biological agents with conventional chemotherapy has led to a significant improvement in response rate, progression-free survival and overall survival in first-line as well as in second- or third-line treatment of MCRC. However, the high variability of response and outcome in MCRC patients treated with these anti-EGFR mAbs has highlighted the need of identifying clinical and/or molecular predictive markers to ensure appropriate use of targeted therapies. The presence of somatic KRAS mutations has been clearly identified as a predictive marker of resistance to anti-EGFR in MCRC, and the use of anti-EGFR mAbs is now restricted to patients with no detectable KRAS mutation. Several studies have indicated that amplification of EGFR, overexpression of the EGFR ligands and inactivation of the anti-oncogene TP53 are associated with sensitivity to anti-EGFR mAbs, whereas mutations of BRAF and PIK3CA and loss of PTEN expression are associated with resistance. Besides these somatic variations, germline polymorphisms such as those affecting genes involved in the EGFR pathway or within the immunoglobulin receptors may also modulate response to anti-EGFR mAbs. Until now, all these markers are not completely validated and only KRAS genotyping is mandatory in routine practice for use of the anti-EGFR mAbs in MCRC.
Similar content being viewed by others


Main
Colorectal cancer (CRC) is the third most common cancer worldwide. Synchronous metastases are estimated to occur in 25% of patients, and ∼40–50% of patients with newly diagnosed CRC develop secondary metastases (Meyerhardt and Mayer, 2005). Since 2004, numerous studies have demonstrated the efficiency of monoclonal antibodies (mAbs) targeting the epidermal growth factor receptor (EGFR) ectodomain, such as cetuximab and panitumumab, in patients with metastatic colorectal cancer (MCRC). Cetuximab corresponds to a chimeric mouse human IgG1 mAb, and panitumumab is a fully human IgG2 mAb.
Indeed, several randomised trials conducted in chemorefractory as well as in chemonaive MCRC patients have reported significant results for cetuximab and panitumumab monotherapy or chemotherapy-based (CT) regimens (Cunningham et al, 2004; Jonker et al, 2007; Van Cutsem et al, 2007, 2009a; Bokemeyer et al, 2009). In a randomised phase II trial including irinotecan-refractory MCRC patients, Cunningham et al (2004) reported that cetuximab plus irinotecan significantly improved the response rate and progression-free survival (PFS) when compared with cetuximab alone (22.9 vs 10.8% and 4.1 vs 1.5 months, respectively). Recently, a phase III randomised trial conducted by Van Cutsem et al (2009a, 2009b) showed that in chemonaive MCRC patients, the addition of anti-EGFR to irinotecan-based CT lead to an 8.2% increase in the objective response (46.8 vs 38.4%), a 0.9-month increase in the PFS (8.9 vs 8 months) and a 1.3-month increase in the overall survival (OS) (19.9 vs 18.6 months). Although the response to anti-EGFR mAbs observed in some patients has confirmed that EGFR activation is oncogenic, as predicted by cellular and animal models, the molecular mechanisms underlying EGFR activation in colorectal cancer remain obscure and are probably heterogeneous. This situation contrasts with in lung adenocarcinoma in which the key mechanism of EGFR activation underlying sensitivity to EGFR inhibitors corresponds to activating mutations within the EGFR tyrosine kinase domain. Although the use of anti-EGFR mAbs was initially restricted to MCRC patients with a detectable expression of EGFR by immunochemistry (IHC), the lack of IHC predictive value and the heterogeneous clinical response have highlighted the need to identify reliable markers predictive of response to anti-EGFR mAbs (Chung et al, 2005).
Two types of molecular predictive markers have been investigated. The majority of studies published so far have analysed somatic alterations affecting effectors of EGFR pathways, such as the receptor–ligand complex, the RAS–RAF–mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3 kinase (PI3K)–Akt–PTEN transduction cascades and p53 (Moroni et al, 2005; Lièvre et al, 2006, 2008; Benvenuti et al, 2007; Di Fiore et al, 2007; Frattini et al, 2007; Khambata-Ford et al, 2007; Amado et al, 2008; De Roock et al, 2008; Di Nicolantonio et al, 2008; Oden-Gangloff et al, 2009; Van Cutsem et al, 2009a). Studies have indicated that amplification of EGFR and overexpression of the EGFR ligands are associated to sensitivity to anti-EGFR mAbs, whereas mutations of BRAF, of PIK3CA and loss of PTEN expression are associated with resistance (Figure 1, bottom panel). The second type of predictive markers investigated are germline polymorphisms of genes involved in the EGFR pathway (Khambata-Ford et al, 2007; Graziano et al, 2008; Garm Spindler et al, 2009) or within the immunoglobulin receptors, considering the potential role of antibody-dependent cellular cytotoxicity (ADCC) in the action of anti-EGFR mAbs (Zhang et al, 2007; Bibeau et al, 2009). At the present time, the only molecular marker predictive of the response to anti-EGFR mAbs, which has been unambiguously validated in MCRC by numerous studies, is the somatic mutation of KRAS as a marker of resistance to anti-EGFR (Lièvre et al, 2006, 2008; Di Fiore et al, 2007; Frattini et al, 2007; Amado et al, 2008; De Roock et al, 2008; Van Cutsem et al, 2009a). This has led to systematic KRAS genotyping in MCRC patients and to the restriction of anti-EGFR mAbs to patients with no detectable KRAS mutation. Nevertheless, KRAS mutations are obviously not the only determinants of the clinical response to anti-EGFR.

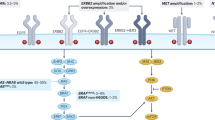
An overview of the EGFR pathway and its main downstream effectors (top). Expected outcomes of anti-EGFR (mAb) therapy (bottom): sensitivity (tumour response) when EGFR is activated (gain copy number, ligand overexpression, other unknown mechanisms) and downstream effectors are wild type (left); resistance (tumour development and metastasis) when downstream effectors such as KRAS, BRAF or PI3K are activated or PTEN is inactivated (right).
Overview of the EGFR pathway
The receptors of EGF are composed of homodimers or heterodimers of four related glycoproteins: HER1 (or EGFR), HER2 (or Erbb2), HER3 and HER4 (Figure 1, top panel). These receptors are composed of an extracellular ligand-binding domain, a transmembrane segment and an intracellular protein tyrosine kinase domain. In a normal cell, activation of EGFR is induced by the binding of the ligands to the ectodomain (Ciardiello and Tortora, 2001, 2008). Approximately ten ligands can activate the EGFR pathway. The ligands for HER1/EGFR are EGF, TGF-α, HB-EGF, amphiregulin, epiregulin and VGF; heregulins are the ligands of Her-3; NRG2, NRG3, heregulins and β-cellulin are the ligands of ErbB-4, and Her-2 is an orphan receptor. Binding of the ligands will induce EGFR homodimerisation (EGFR-EGFR) or heterodimerisation (EGFR-HER2; EGFR-HER3 or EGFR-HER4) and then ATP-dependent phosphorylation of tyrosine residues located within the intracellular domain. The phosphorylated EGFR will then lead to the activation of the RAS–RAF–MAPK kinase and the PI3K–Akt transduction cascades. The RAS–RAF–MAPK kinase is the major downstream signalling route of the EGFR pathway and it controls cell-cycle progression, differentiation, survival and in particular, the G1/S-phase transition (Ciardiello and Tortora, 2008). The RAS proteins are members of a large superfamily of guanine guanosine-5′-triphosphate (GTP) and guanine guanosine-5′-diphosphate (GDP)-binding proteins. In normal cells, EGFR activates RAS by stimulating its binding to GTP. RAF will then be activated and will phosphorylate the MAP2K1 and MAP2K2 kinases. The p110 subunit of PI3K encoded by the PIK3CA oncogene is activated by RAS proteins. The PI3K–Akt pathway, which is negatively regulated by the PTEN protein, activates antiapoptotic and survival signals (Figure 1, top panel).
In cancerous cells, EGFR pathway activation results in cell proliferation, inhibition of apoptosis, activation of invasion, metastasis and tumour neovascularisation (Ciardiello and Tortora, 2001, 2008; Cohen et al, 2005). The main mechanism of EGFR activation, so far characterised in CRC, corresponds to the somatic mutation of the KRAS proto-oncogene. Somatic mutations of KRAS are detected in ∼30–40% of CRCs (Andreyev et al, 2001). These activating mutations are missense mutations that introduce amino-acid substitutions mainly at positions 12, 13 and 61. Activating mutations of other EGFR pathway effectors have also been described. In CRC, mutations of BRAF and PI3K genes are detected in ∼5–10% and 6–13% of the tumours, respectively (Moroni et al, 2005; Lièvre et al, 2006; Di Nicolantonio et al, 2008; Laurent-Puig et al, 2009; Prenen et al, 2009; Sartore-Bianchi et al, 2009). Somatic mutations of KRAS and BRAF are mutually exclusive.
KRAS mutation: a validated predictive marker of resistance to anti-EGFR
Since 2004, the predictive value of somatic KRAS mutation, in terms of resistance to anti-EGFR mAbs, has been established by numerous studies. These studies (see Table 1), mainly focusing on mutations of codons 12 and 13, and more recently on codon 61, have been based on molecular analyses of tumour-extracted DNAs from patients included in retrospective studies, as well as in prospective randomised trials (Lièvre et al, 2006, 2008; Benvenuti et al, 2007; Di Fiore et al, 2007; Frattini et al, 2007; Amado et al, 2008; De Roock et al, 2008; Bokemeyer et al, 2009; Loupakis et al, 2009a; Van Cutsem et al, 2009a). The first line of evidence that KRAS mutation was a strong predictor of resistance to anti-EGFR mAbs was the observation that in chemorefractory patients treated with cetuximab-based CT, <2% of patients with detectable KRAS mutation exhibited an objective response, whereas ∼40% of patients with no detectable KRAS mutation showed a clinical response (Lièvre et al, 2006, 2008; Benvenuti et al, 2007; Di Fiore et al, 2007, 2008a, 2008b; Frattini et al, 2007; De Roock et al, 2008; Loupakis et al, 2009a). The impact of KRAS status in response to anti-EGFR mAbs was also documented by trials in chemorefractory patients comparing cetuximab or panitumumab monotherapy vs best supportive care, with an increase in the response rate in patients with no KRAS mutation from 8 to 12.8% and from 10 to 17%, respectively (Karapetis et al, 2008).
In chemonaive patients with no detectable KRAS mutation, the addition of anti-EGFR antibodies to CT increased the objective response from approximately 46 to 60% when compared with CT alone (Douillard et al, 2008, 2009; Bokemeyer et al, 2009; Van cutsem et al, 2009a). Except in the recent COIN randomised trial (Maughan et al, 2009), the addition of anti-EGFR mAbs to CT has also been shown to lead to a significant increase in the median PFS when compared with irinotecan- or oxaliplatin-based CT alone in chemonaive as well as in chemorefractory patients. In contrast, in mutant KRAS patients, no clinical benefit was observed from the combination of anti-EGFR plus CT. The effect of KRAS status on OS in MCRC patients treated in first line remains debatable, with contradictory results that are mainly explained by the non-standardisation of second- and third-line CT as well as several confounding factors such as treatment crossover and different monitoring methods used in these such randomised trials (Douillard et al, 2009; Van Cutsem et al, 2009a, 2009b).
Other somatic predictive markers of response to anti-EGFR
PIK3CA and PTEN alterations
The demonstration that in MCRC patients KRAS mutations confer resistance to anti-EGFR has validated the hypothesis that targeting EGFR will be inefficient if activation of the EGFR transduction cascade results from somatic alterations affecting downstream effectors (Figure 1, bottom panel). This led to an investigation of the predictive value of somatic alterations affecting other effectors. In colorectal cancer, activation of the PIK3CA pathway can result either from activating PIK3CA mutations or from inactivation of the PTEN protein. Since 2005, the impact of PIK3CA mutations on MCRC patients treated with anti-EGFR has been investigated by several studies that have reported a PIK3CA mutation frequency ranging from 6 to 13% (Moroni et al, 2005; Lièvre et al, 2006; Perrone et al, 2009; Prenen et al, 2009; Sartore-Bianchi et al, 2009). In the studies of Moroni et al (2005), Lièvre et al (2006) and Perrone et al (2009), none of the responders to anti-EGFR therapy had a PIK3CA mutation, but this association was not statistically significant, given the limited number of patients. In contrast, a significant association between the presence of PIK3CA mutation and response to anti-EGFR has been recently reported by Sartore-Bianchi et al (2009) in a study including 110 patients treated with anti-EGFR mAbs. A total of 32 (29%) patients had a KRAS mutation and 15 (13%) had a PIK3CA mutation. When compared with the unselected population, the response rate to anti-EGFR increased from 20 to 26% in patients with no KRAS mutation, to 23% in patients with no PIK3CA mutation and to 31% in patients with no KRAS/PIK3CA mutations (Sartore-Bianchi et al, 2009). Nevertheless, these results were not confirmed by the recent study of Prenen et al (2009), in which PIK3CA mutations were identified in 5 of 39 (13%) responders vs 18 of 160 (11%) nonresponders (P=0.781). These contradictory results highlight the fact that the association between PIK3CA mutations and resistance to anti-EGFR is not yet validated.
PTEN exerts an effect as a tumour suppressor by dephosphorylating the plasma membrane lipid second messenger PIP-3 generated by the action of PI3KCA. The loss of PTEN function induces an increase in PIP-3 concentration and PIK3CA pathway activation. IHC and FISH analyses are the two main methods that have been used to evaluate the PTEN status in studies focused on anti-EGFR resistance. However, the molecular mechanisms leading to PTEN inactivation are heterogeneous and include genomic deletions, inactivating mutations and promoter hypermethylation, indicating that PTEN integrity cannot be explored by a simple method. In a series of 27 MCRC patients, Frattini et al (2007) reported that PTEN alteration detected by IHC in primary tumours was significantly associated with cetuximab resistance. More recently, Loupakis et al (2009b) have analysed by IHC the impact of PTEN cytoplasmic staining intensity in 85 primary tumours and 55 related metastases from patients treated with cetuximab plus irinotecan. First, using a specific scoring system for PTEN expression, these authors found that IHC staining was concordant in only 27 out of 45 (60%) pairs of assessable primary and related metastases. Second, the results of the PTEN IHC performed on the 85 primary tumours did not correlate with the response and PFS, whereas PTEN IHC analysis performed on the 55 metastases was significantly associated with response rate and PFS. When the KRAS mutational status in the primary tumours and the PTEN expression in metastasis were combined, the subgroup of patients with no detectable KRAS mutation and positive to PTEN had a significantly higher response and longer PFS than patients with KRAS mutation and negative to PTEN. In patients with no KRAS mutation, Laurent-Puig et al (2009) found that the absence of PTEN expression, detected in 19.9% of patients using another IHC scoring system, was associated with shorter OS in univariate and multivariate analyses. In a series of 72 MCRC patients treated with anti-EGFR mAbs plus CT, Razis et al (2008) evaluated the correlation of PTEN gene copy number and sensitivity to anti-EGFR. The absence of PTEN gene dosage alteration was significantly associated with response rate, whereas PTEN gene deletion was correlated with worse PFS. No significant association was found using PTEN IHC expression. All these results clearly indicate that no obvious association has yet been established between PTEN alterations and anti-EGFR therapy in MCRC patients. It should also be noted that commercially available anti-PTEN antibodies do not always provide reproducible results in IHC. Further studies are needed to determine the most efficient and reproducible methods for PI3KCA and PTEN analysis in order to clearly evaluate the predictive value of each marker separately or in combination with KRAS status.
BRAF mutation
Two studies support that V600E mutation, resulting in strong activation of the BRAF protein downstream to KRAS, was associated with shorter PFS and OS in MCRC chemorefractory patients treated with anti-EGFR mAbs (Di Nicolantonio et al, 2008; Laurent-Puig et al, 2009). Nevertheless, it seems that whatever the treatment, this mutation in MCRC is mainly associated with poor prognosis, which may interfere with its predictive value regardless of anti-EGFR mAbs (Tol et al, 2010; Van Cutsem et al, 2010).
TP53 mutations
In a normal cell, the p53 protein exerts an effect not only as a guardian of the genome, which is activated when DNA damage occurs, but also as a policeman of oncogenes, which become active when oncogenes are inappropriately activated, thus inducing apoptosis and/or senescence (Efeyan and Serrano, 2007; Halazonetis et al, 2008). Alteration of the p53 pathway has been reported to be systematically observed in non-small cell lung cancer with activating EGFR mutations, suggesting that p53 inactivation is required to allow expansion of a cell with EGFR pathway activation (Mounawar et al, 2007). On the other hand, it has been shown that PI3K signalling activates p53-mediated growth suppression, suggesting that p53 exerts an effect as a brake for the activated PI3K transduction cascade (Kim et al, 2007). Taken together, these data led us to formulate the hypothesis that activation of the EGFR pathway should be oncogenic, and therefore anti-EGFR antibodies should only be efficient in tumours, only if p53 is inactivated. We have recently analysed the impact of TP53 mutations on 64 MCRC patients treated with cetuximab plus CT (Oden-Gangloff et al, 2009). In this series, TP53 mutations were found in 41 of 64 patients and were significantly associated with controlled disease (P=0.037) and higher PFS (20 vs 12 weeks, P=0.004). In the subgroup of patients without KRAS detectable mutation, we have also found that controlled disease and PFS were significantly improved in TP53 mutated patients when compared with TP53 nonmutated patients. These results might be explained not only by the fact that EGFR activation is oncogenic only if TP53 is inactivated, but also by the fact that inactivation of TP53 could be one of the mechanisms leading to EGFR activation. Cellular models and studies on larger MCRC series are necessary to clarify the relationship between TP53 status and sensitivity to anti-EGFR.
EGFR gene copy number and expression of its ligands
EGFR gene copy number
A common mechanism of EGFR activation is the increase in the EGFR copy number corresponding to the gain of copies or amplification of the chromosome 7 region on which EGFR is located. A copy number increase of EGFR resulting from chromosome 7 polysomy probably does not have the same biological significance than amplification of the EGFR locus. In contrast to breast cancer in which tumour cells exhibit HER2 amplification in homogeneous regions with a relationship between expression and gene amplification, colorectal cancer is characterised by a heterogeneous EGFR gene copy number pattern and, furthermore, no correlation has been established between the EGFR copy number and the EGFR IHC staining. Based on the results obtained in patients with metastatic breast cancer treated with trastuzumab, in which ERBB2 amplification correlates with sensitivity to mAb therapy, Moroni et al (2005) investigated the impact of the EGFR gene copy number, assessed by FISH, on the anti-EGFR mAb response in a series of 30 MCRC patients. This study reported a positive significant association between an increased EGFR gene copy number and the response rate to cetuximab or panitumumab. Since 2005, many reports have investigated the relationship between EGFR copy number and response to mAb therapy in MCRC, but only five of them restricted the analysis to the subgroup of wild-type KRAS patients (Table 2). Although an increase in EGFR copy number is significantly associated with mAb therapy response, the results are less obvious in terms of PFS or OS. Cappuzzo et al (2008), in a series of 85 chemorefractory patients treated with cetuximab, determined that the best cutoff significantly associated with the response rate and PFS was 2.92 EGFR gene copy number per cell. Interestingly, these authors further analysed their population according to the EGFR copy number cutoff previously reported by Sartore-Bianchi et al (2009) and observed a significant association for response rate but not for PFS. It can be concluded from the different studies published so far that only a high EGFR gene copy number, resulting from gene amplification and observed in a small fraction of patients, is probably predictive of anti-EGFR sensitivity in MCRC patients.
Expression of EGFR ligands
Few studies have investigated the predictive role of EGFR ligand expression on anti-EGFR response in MCRC. A transcriptome analysis of 164 CRCs, using a 640 probe set, showed that the EGFR ligands, epiregulin (EREG) and amphiregulin (AREG), were highly expressed in 25% of the samples (Khambata-Ford et al, 2007). In this study, analysis of metastatic frozen samples from a population of 80 MCRC patients treated with cetuximab monotherapy showed that patients with disease control expressed the EGFR ligands at a higher level than nonresponders. Moreover, patients with tumours exhibiting higher EREG or AREG expression had significantly longer PFS, suggesting that these tumours were EGFR dependent and were therefore particularly sensitive to the inhibition of the ligand–receptor interaction by anti-EGFR mAb (Khambata-Ford et al, 2007). In a recent study performed on paraffin-embedded samples in refractory MCRC patients treated with cetuximab and irinotecan, the KRAS status and the EREG and AREG mRNA expression were first determined on 220 primary samples and the results were then validated on a series of 67 patients (Jacobs et al, 2009). The mRNA levels of EREG and AREG significantly correlated with the absence of detectable KRAS mutation. When ligand expression level was combined with the KRAS status, the median OS was 65 weeks in patients with no detectable KRAS mutation and a high EREG expression vs 31 weeks (P=0.01) in patients without detectable KRAS mutation and low EREG expression (Jacobs et al, 2009).
Germline polymorphisms associated with clinical response to anti-EGFR mAbs
Three types of germline polymorphisms suspected of modulating the response to anti-EGFR have been investigated. The first corresponds to a CA repeat present within the EGFR intron 1, which has been shown to modulate EGFR transcription efficiency, the longer (L) repeats being associated with a reduced transcription when compared with the shorter (S) ones. The second, located within the EGF gene, corresponds to a SNP (G>A, rs4444903). In 110 chemorefractory MCRC patients treated with cetuximab–irinotecan, Graziano et al (2008) showed that EGFR intron-1 variant (S/S vs L/L and L/S) and EGF61 variant (G/G vs A/A and A/G) were significantly associated with a better OS. Similar results were obtained on EGF61 genotypes in a study on chemorefractory MCRC patients treated with cetuximab plus irinotecan CT (Garm Spindler et al, 2009). In patients with no detectable KRAS mutation, homozygous phenotypes (A/A and G/G) were associated with a lower progression rate (19 vs 60%) than EGF61 A/G patients (P=0.006) and a significant increase in OS (17.1 vs 5.9 months). The third type of germline polymorphism investigated, in the context of anti-EGFR therapies in MCRC, corresponds to polymorphic amino-acid substitutions within the receptors of the Fc fragment of the immunoglobulins (Igs). The H131R and V158F variations within the Fcγ RIIa and Fcγ RIIIa have been shown to modify the affinity of the Ig receptors and are therefore predicted to modulate the ADCC in patients receiving anti-EGFR IgG1 mAb. To date, only four studies have found significant association between these polymorphisms and the clinical outcome. In a first study performed in a series of 39 chemorefractory patients treated with cetuximab monotherapy, Zhang et al (2007) found that the presence of the FCGR2A 131 H allele and of the FCGR3A 158 F allele were significantly associated with better PFS; however, these results could not be replicated on a larger series including 130 subjects (Lurje et al, 2008). In 64 chemorefractory MCRC patients treated with cetuximab-based CT, Bibeau et al (2009) found that the FCGR2A 131 H/H and FCGR3A 158 V/V genotypes, predicted to result in a higher affinity of the Ig to the receptor, were significantly associated with better PFS. In this study, analysis of these germline polymorphisms in the subgroup of patients with no detectable KRAS mutation showed that the median PFS was 9.6 months in the 131 H/H or 158 V/V patients vs 4.6 months in the subgroup of patients harbouring the other genotypes (P=0.015). In contrast, a more recent study (Pander et al, 2010), performed on KRAS wild-type patients receiving chemotherapy plus cetuximab, showed that subjects homozygous for the FCGR3A 158 F allele have a better PFS than those bearing other allelic combinations. Several factors may account for these discrepancies; although ethnic origin is unlikely to be involved, previous treatment regimens and association of irinotecan with cetuximab could modify patients’ response. Further studies involving testing integrity of the EGFR pathway are clearly needed to clarify this point.
Mutational status in primary colorectal adenocarcinomas and related metastases
One of the main subjects of debate is the relevance of KRAS genotyping on primary tumours, whereas anti-EGFR mAbs are used to treat a metastatic disease. Indeed, some publications have indicated that concordance of mutational status of KRAS and other EGFR downstream effectors between primary and metastases is not absolute. In a series of 48 CRC patients, an overall concordance of 92% was observed between primary tumours and metastases for the presence of mutations within KRAS exon 2 and BRAF exon 5 (Artale et al, 2008); however, in the specific subgroup of patients with KRAS mutation, 23% of discordance was observed between the two tumour sites. In a study including 38 MCRCs, Molinari et al (2009) reported a concordance for KRAS and BRAF mutational status of 92 and 100%, respectively. More recently, the analysis of KRAS, BRAF and PIK3CA in tumour invasion fronts, lymph nodes and distant metastases revealed a discordance between primary tumours and lymph node metastases of 31, 4 and 13%, respectively (Baldus et al, 2010). A discordance between primary tumours and distant metastases was observed for KRAS in 10% of the cases and for PIK3CA in 5% of the cases (Baldus et al, 2010). In a large series of >800 samples tested for KRAS, using a highly sensitive SNaPshot-based method (Di Fiore et al, 2007), we observed a discordant KRAS mutational status between tumours and metastases in 10% of the paired samples (Lamy et al, 2010). These studies clearly indicate that given the genetic heterogeneity and genetic evolution of CRC under treatment, the most appropriate tissue on which molecular profiling should be performed remains to be determined.
Conclusion and perspectives
In conclusion, the numerous studies published since 2006 on the markers of sensitivity and resistance to anti-EGFR mAbs in MCRC have clearly validated somatic KRAS mutations as markers of resistance to anti-EGFR, resulting in restricted use of these targeted therapies to patients with no detectable KRAS mutation. However, in these patients, the heterogeneity of response strongly suggests that several genetic variations, corresponding either to somatic alterations present only within the tumours or to germline polymorphisms, can modulate sensitivity or resistance to anti-EGFR mAbs. One of the challenges is now to identify these other genetic predictive markers, using either candidate gene approaches, global analysis based on pangenomic/transcriptomic/proteomic portraits or cellular or animal models. Some technical and conceptual questions concerning KRAS genotyping remain unsolved. One important question under debate is to define in MCRC patients, the most appropriate tissue on which DNA analysis should be performed in the context of targeted therapies. Indeed, most studies published so far are based on the analysis of genetic material extracted from primary tumours, whereas targeted therapies are mainly directed at metastatic disease treatment. As highlighted by several recent studies, this approach may have limitations: (1) the primary tumour may not always be available; (2) as indicated above, several genetic changes may occur between the primary tumour and the corresponding metastases; and (3) genetic analyses performed on paraffin-embedded tissues may generate artefacts because of fragmentation and chemical modification of DNA. Therefore, we think, as previously shown, that genetic analysis based on highly sensitive detection from blood of circulating mutant DNA might be of clinical interest (Di Fiore et al, 2008a; Yen et al, 2009).
As the new markers will have to be validated by independent studies, the challenge for the clinician, before treating patients, will be the progressive integration of these markers in the routine and the development of a decisional algorithm, which should probably be stratified first by KRAS genotyping. In patients with KRAS mutations, the choice of the most appropriate treatment is problematic. Although the use of anti-EGFR mAbs is not recommended in these patients, it should be underlined that disease stability can be observed in up to 50% of chemorefractory patients treated with anti-EGFR alone in association with CT (Di Fiore et al, 2008b). In these patients, targeted therapies will probably be based, in the future, on molecules able to inhibit effectors of the EGFR transduction cascade and located downstream from the KRAS protein.
Change history
29 March 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Andreyev HJ, Norman AR, Cunningham D, Oates J, Dix BR, Iacopetta BJ, Young J, Walsh T, Ward R, Hawkins N, Beranek M, Jandik P, Benamouzig R, Jullian E, Laurent-Puig P, Olschwang S, Muller O, Hoffmann I, Rabes HM, Zietz C, Troungos C, Valavanis C, Yuen ST, Ho JW, Croke CT, O’Donoghue DP, Giaretti W, Rapallo A, Russo A, Bazan V, Tanaka M, Omura K, Azuma T, Ohkusa T, Fujimori T, Ono Y, Pauly M, Faber C, Glaesener R, de Goeij AF, Arends JW, Andersen SN, Lövig T, Breivik J, Gaudernack G, Clausen OP, De Angelis PD, Meling GI, Rognum TO, Smith R, Goh HS, Font A, Rosell R, Sun XF, Zhang H, Benhattar J, Losi L, Lee JQ, Wang ST, Clarke PA, Bell S, Quirke P, Bubb VJ, Piris J, Cruickshank NR, Morton D, Fox JC, Al-Mulla F, Lees N, Hall CN, Snary D, Wilkinson K, Dillon D, Costa J, Pricolo VE, Finkelstein SD, Thebo JS, Senagore AJ, Halter SA, Wadler S, Malik S, Krtolica K, Urosevic N (2001) Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. Br J Cancer 85: 692–696
Amado GR, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, Juan T, Sikorski R, Suggs S, Radinsky R, Patterson SD, Chang DD (2008) Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 26: 1626–1634
Artale S, Sartore-Bianchi A, Veronese SM, Gambi V, Sarnataro CS, Gambacorta M, Lauricella C, Siena S (2008) Mutations of KRAS and BRAF in primary and matched metastatic sites of colorectal cancer. J Clin Oncol 26: 4217–4219
Baldus SE, Schaefer KL, Engers R, Hartleb D, Stoecklein NH, Gabbert HE (2010) Prevalence and heterogeneity of KRAS, BRAF, and PIK3CA mutations in primary colorectal adenocarcinomas and their corresponding metastases. Clin Cancer Res 16: 790–799
Benvenuti S, Sartore-Bianchi A, Di Nicolantonio F, Zanon C, Moroni M, Veronese S, Siena S, Bardelli A (2007) Oncogenic activation of the RAS/RAF signaling pathway impairs the response of metastatic colorectal cancers to anti-epidermal growth factor receptor antibody therapies. Cancer Res 67: 2643–2648
Bibeau F, Lopez-Crapez E, Di Fiore F, Thezenas S, Ychou M, Blanchard F, Lamy A, Penault-Llorca F, Frébourg T, Michel P, Sabourin JC, Boissière-Michot F (2009) Impact of Fc{gamma}RIIa-Fc{gamma}RIIIa polymorphisms and KRAS mutations on the clinical outcome of patients with metastatic colorectal cancer treated with cetuximab plus irinotecan. J Clin Oncol 27: 1122–1129
Bokemeyer C, Bondarenko I, Makhson A, Hartmann JT, Aparicio J, de Braud F, Donea S, Ludwig H, Schuch G, Stroh C, Loos AH, Zubel A, Koralewski P (2009) Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J Clin Oncol 27: 663–671
Bokemeyer C, Bondarenko I, Hartmann JT, De Braud FG, Schuch G, Zubel A, Celik I, Koralewski P (2010) Biomarkers predictive for outcome in patients with metastatic colorectal cancer (mCRC) treated with first-line FOLFOX4 plus or minus cetuximab: Updated data from the OPUS study. Gastrointestinal Cancer Symposium, Abstract 428
Cappuzzo F, Finocchiaro G, Rossi E, Jänne PA, Carnaghi C, Calandri C, Bencardino K, Ligorio C, Ciardiello C, Pressiani T, Destro A, Roncalli M, Crino L, Franklin WA, Santoro A, Varella-Garcia M (2008) EGFR FISH assay predicts for response to cetuximab in chemotherapy refractory colorectal cancer patients. Ann Oncol 19: 717–723
Chung KY, Shia J, Kemeny NE, Shah M, Schwartz GK, Tse A, Hamilton A, Pan D, Schrag D, Schwartz L, Klimstra DS, Fridman D, Kelsen DP, Saltz LB (2005) Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J Clin Oncol 23: 1803–1810
Ciardiello F, Tortora G (2008) EGFR antagonists in cancer treatment. N Engl J Med 358: 1160–1174
Ciardiello F, Tortora G (2001) A novel approach in the treatment of cancer: targeting the epidermal growth factor receptor. Clin Cancer Res 7: 2958–2970
Cohen SJ, Cohen RB, Meropol NJ (2005) Targeting signal transduction pathways in colorectal cancer – more than skin deep. J Clin Oncol 23: 5374–5385
Cunningham D, Humblet Y, Siena S, Khayat D, Bleiberg H, Santoro A, Bets D, Mueser M, Harstrick A, Verslype C, Chau I, Van Cutsem E (2004) Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 351: 337–345
De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, Biesmans B, Van Laethem JL, Peeters M, Humblet Y, Van Cutsem E, Tejpar S (2008) KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol 19: 508–515
Di Fiore F, Blanchard F, Charbonnier F, Le Pessot F, Lamy A, Galais MP, Bastit L, Killian A, Sesboüé R, Tuech JJ, Queuniet AM, Paillot B, Sabourin JC, Michot F, Michel P, Frebourg T (2007) Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Br J Cancer 96: 1166–1169
Di Fiore F, Charbonnier F, Lefebure B, Laurent M, Le Pessot F, Michel P, Frebourg T (2008a) Clinical interest of KRAS mutation detection in blood for anti-EGFR therapies in metastatic colorectal cancer. Br J Cancer 99: 551–552
Di Fiore F, Van Cutsem E, Laurent-Puig P, Siena S, Frattini M, De Roock W, Lièvre A, Sartore-Bianchi A, Bardelli A, Tejpar S (2008b) Role of KRAS mutation in predicting response, progression-free survival, and overall survival in irinotecan-refractory patients treated with cetuximab plus irinotecan for a metastatic colorectal cancer: analysis of 281 individual data from published series. J Clin Oncol 26 (Suppl): Abstract 4035
Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A (2008) Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 26: 5705–5712
Douillard J, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel ME, Humblet Y, Cunningham D, Wolf M, Gansert JL (2009) Randomized phase 3 study of panitumumab with FOLFOX4 compared to FOLFOX4 alone as 1st-line treatment (tx) for metastatic colorectal cancer (mCRC): the PRIME trial. ECCO-ESMO LBA10. Eur J Cancer Supp, 7: 6
Efeyan A, Serrano M (2007) P53: guardian of the genome and policeman of the oncogenes. Cell Cycle 6: 1006–1010
Frattini M, Saletti P, Romagnani E, Martin V, Molinari F, Ghisletta M, Camponovo A, Etienne LL, Cavalli F, Mazzucchelli L (2007) PTEN loss of expression predicts cetuximab efficacy in metastatic colorectal cancer patients. Br J Cancer 97: 1139–1145
Garm Spindler KL, Pallisgaard N, Rasmussen AA, Lindebjerg J, Andersen RF, Crüger D, Jakobsen A (2009) The importance of KRAS mutations and EGF61A>G polymorphism to the effect of cetuximab and irinotecan in metastatic colorectal cancer. Ann Oncol 20: 879–884
Graziano F, Ruzzo A, Loupakis F, Canestrari E, Santini D, Catalano V, Bisonni R, Torresi U, Floriani I, Schiavon G, Andreoni F, Maltese P, Rulli E, Humar B, Falcone A, Giustini L, Tonini G, Fontana A, Masi G, Magnani M (2008) Pharmacogenetic profiling for cetuximab plus irinotecan therapy in patients with refractory advanced colorectal cancer. J Clin Oncol 26: 1427–1434
Halazonetis TD, Gorgoulis VG, Bartek J (2008) An oncogene-induced DNA damage model for cancer development. Science 319: 1352–1355
Jacobs B, De Roock W, Piessevaux H, Van Oirbeek R, Biesmans B, De Schutter J, Fieuws S, Vandesompele J, Peeters M, Van Laethem JL, Humblet Y, Penault-Llorca F, De Hertogh G, Laurent-Puig P, Van Cutsem E, Tejpar S (2009) Amphiregulin and epiregulin mRNA expression in primary tumors predicts outcome in metastatic colorectal cancer treated with cetuximab. J Clin Oncol 27: 5068–5074
Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Dongsheng T, Au HJ, Berry SR, Krahn M, Price T, Simes RJ, Tebutt NC, Van Hazel G, Wierzbicki R, Langer C, Moore MJ (2007) Cetuximab for the treatment of colorectal cancer. N Engl J Med 357: 2040–2048
Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC, Simes RJ, Chalchal H, Shapiro JD, Robitaille S, Price TJ, Shepherd L, Au HJ, Langer C, Moore MJ, Zalcberg JR (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359: 1757–1765
Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, Wong TW, Huang X, Takimoto C, Godwin AK, Tan BR, Krishnamurthi SS, Burris III HA, Poplin EA, Hidalgo M, Baselga J, Clark EA, Mauro DJ (2007) Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol 25: 3230–3237
Kim JS, Lee C, Bonifant C, Ressom H, Waldman T (2007) Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol Cell Biol 27: 662–677
Lamy A, Blanchard F, Le Pessot F, Houlle S, Bossut J, Di Fiore F, Sesboüé R, Sabourin JC (2010) Metastatic CRCs KRAS genotyping in routine diagnosis: Results and pitfalls. ASCO GI Abstract 294
Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T, Rougier P, Lievre A, Landi B, Boige V, Ducreux M, Ychou M, Bibeau F, Bouché O, Reid J, Stone S, Penault-Llorca F (2009) Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol 27: 5924–5930
Lièvre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF, Côté JF, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P (2006) KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 66: 3992–3995
Lièvre A, Bachet JB, Boige V, Cayre A, Le Corre D, Buc E, Ychou M, Bouché O, Landi B, Louvet C, André T, Bibeau F, Diebold MD, Rougier P, Ducreux M, Tomasic G, Emile JF, Penault-Llorca F, Laurent-Puig P (2008) KRAS mutation as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J Clin Oncol 26: 374–379
Loupakis F, Pollina L, Stasi I, Ruzzo A, Scartozzi M, Santini D, Masi G, Graziano F, Cremolini C, Rulli E, Canestrari E, Funel N, Schiavon G, Petrini I, Magnani M, Tonini G, Campani D, Floriani I, Cascinu S, Falcone A (2009b) PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J Clin Oncol 27: 2622–2629
Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D, Masi G, Stasi I, Canestrari E, Rulli E, Floriani I, Bencardino K, Galluccio N, Catalano V, Tonini G, Magnani M, Fontanini G, Basolo F, Falcone A, Graziano F (2009a) KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer 101: 715–721
Lurje G, Nagashima F, Zhang W, Yang D, Chang HM, Gordon MA, El-Khoueiry A, Husain H, Wilson PM, Ladner RD, Mauro DJ, Langer C, Rowinsky EK, Lenz HJ (2008) Polymorphisms in cyclooxygenase-2 and epidermal growth factor receptor are associated with progression-free survival independent of K-ras in metastatic colorectal cancer patients treated with single-agent cetuximab. Clin Cancer Res 14: 7884–7895
Maughan T, Adams RA, Smith CG, Seymour MT, Wilson R, Meade AM, Fisher D, Madi A, Cheadle J, Kaplan R (2009) Addition of cetuximab to oxaliplatin-based combination chemotherapy in patients with KRAS wild-type advanced colorectal cancer (ACRC): a randomised superiority trial (MRC COIN). ECCO-ESMO LBA6. Eur J Cancer Supp, 7: 4
Meyerhardt JA, Mayer RJ (2005) Systemic therapy for colorectal cancer. N Engl J Med 352: 476–487
Molinari F, Martin V, Saletti P, De Dosso S, Spitale A, Camponovo A, Bordoni A, Crippa S, Mazzucchelli L, Frattini M (2009) Differing deregulation of EGFR and downstream proteins in primary colorectal cancer and related metastatic sites may be clinically relevant. Br J Cancer 100: 1087–1094
Moroni M, Veronese S, Benvenuti S, Marrapese G, Sartore-Bianchi A, Di Nicolantonio F, Gambacorta M, Siena S, Bardelli A (2005) Gene copy number for epidermal growth factor receptor (EGFR) and clinical response to antiEGFR treatment in colorectal cancer: a cohort study. Lancet Oncol 6: 279–286
Mounawar M, Mukeria A, Le Calvez F, Hung RJ, Renard H, Cortot A, Bollart C, Zaridze D, Brennan P, Boffetta P, Brambilla E, Hainaut P (2007) Patterns of EGFR, HER2, TP53, and KRAS mutations of p14arf expression in non-small cell lung cancers in relation to smoking history. Cancer Res 67: 5667–5672
Oden-Gangloff A, Di Fiore F, Bibeau F, Lamy A, Bougeard G, Charbonnier F, Blanchard F, Tougeron D, Ychou Y, Boissière F, Le Pessot F, Sabourin JC, Tuech JJ, Michel P, Frebourg T (2009) TP53 mutations predict disease control in metastatic colorectal cancer treated with cetuximab-based chemotherapy. Br J Cancer 100: 1330–1335
Pander J, Gelderblom H, Antonini NF, Tol J, Krieken JH, Straaten TV, Punt CJ, Guchelaar HJ (2010) Correlation of FCGR3A and EGFR germline polymorphisms with the efficacy of cetuximab in KRAS wild-type metastatic colorectal cancer. Eur J Cancer 46: 1829–1834
Peeters M, Price T, Hotko Y, Cervantes A, Ducreux M, Andre T, Chan E, Lordick F, Rong A, Gansert J (2009) Randomized phase 3 study of panitumumab with FOLFIRI compared to FOLFIRI alone as second-line treatment (tx) in patients (pts) with metastatic colorectal cancer (mCRC). ECCO-ESMO LBA14. Eur J Cancer Supp, 7: 9
Perrone F, Lampis A, Orsenigo M, Di Bartolomeo M, Gevorgyan A, Losa M, Frattini M, Riva C, Andreola S, Bajetta E, Bertario L, Leo E, Pierotti MA, Pilotti S (2009) PI3KCA/PTEN deregulation contributes to impaired responses to cetuximab in metastatic colorectal cancer patients. Ann Oncol 20: 84–90
Personeni N, Fieuws S, Piessevaux H, De Hertogh G, De Schutter J, Biesmans B, De Roock W, Capoen A, Debiec-Rychter M, Van Laethem JL, Peeters M, Humblet Y, Van Cutsem E, Tejpar S (2008) Clinical usefulness of EGFR gene copy number as a predictive marker in colorectal cancer patients treated with cetuximab: a fluorescent in situ hybridization study. Clin Cancer Res 14: 5869–5876
Prenen H, De Schutter J, Jacobs B, De Roock W, Biesmans B, Claes B, Lambrechts D, Van Cutsem E, Tejpar S (2009) PIK3CA mutations are not a major determinant of resistance to the epidermal growth factor receptor inhibitor cetuximab in metastatic colorectal cancer. Clin Cancer Res 15: 3184–3188
Razis E, Briasoulis E, Vrettou E, Skarlos DV, Papamichael D, Kostopoulos I, Samantas E, Xanthakis I, Bobos M, Galanidi E, Bai M, Gikonti I, Koukouma A, Kafiri G, Papakostas P, Kalogeras KT, Kosmidis P, Fountzilas G (2008) Potential value of PTEN in predicting cetuximab response in colorectal cancer: an exploratory study. BMC Cancer 8: 234
Sartore-Bianchi A, Martini M, Molinari F, Veronese S, Nichelatti M, Artale S, Di Nicolantonio F, Saletti P, De Dosso S, Mazzucchelli L, Frattini M, Siena S, Bardelli A (2009) PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res 69: 1851–1857
Scartozzi M, Bearzi I, Mandolesi A, Pierantoni C, Loupakis F, Zaniboni A, Negri F, Quadri A, Zorzi F, Galizia E, Berardi R, Biscotti T, Labianca R, Masi G, Falcone A, Cascinu S (2009) Epidermal growth factor receptor (EGFR) gene copy number (GCN) correlates with clinical activity of irinotecan-cetuximab in K-RAS wild-type colorectal cancer: a fluorescence in situ (FISH) and chromogenic in situ hybridization (CISH) analysis. BMC Cancer 9: 303
Tol J, Dijkstra JR, Klomp M, Teerenstra S, Dommerholt M, Vink-Börger ME, van Cleef PH, van Krieken JH, Punt CJ, Nagtegaal ID (2010) Markers for EGFR pathway activation as predictor of outcome in metastatic colorectal cancer patients treated with or without cetuximab. Eur J Cancer 46: 1997–2009
Van Cutsem E, Köhne CH, Hitre E, Zaluski J, Chang Chien CR, Makhson A, D’Haens G, Pintér T, Lim R, Bodoky G, Roh JK, Folprecht G, Ruff P, Stroh C, Tejpar S, Schlichting M, Nippgen J, Rougier P (2009a) Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N Engl J Med 360: 1408–1417
Van Cutsem E, Lang I, Folprecht G, Nowacki M, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Celik I, Kohne C (2010) Cetuximab plus FOLFIRI in the treatment of metastatic colorectal cancer (mCRC): the influence of KRAS and BRAF biomarkers on outcome: updated data from the CRYSTAL trial. ASCO GI, Abstract 281
Van Cutsem E, Peeters M, Siena S, Humblet Y, Hendlisz A, Neyns B, Canon JL, Van Laethem JL, Maurel J, Richardson G, Wolf M, Amado RG (2007) Open-label phase III trial of panitumumab plus best supportive care alone in patients with chemotherapy –refractory metastatic colorectal cancer. J Clin Oncol 25: 1658–1664
Van Cutsem E, Rougier P, Köhne C, Stroh C, Schlichting M, Bokemeyer C (2009b) A meta-analysis of the CRYSTAL and OPUS studies combining cetuximab with chemotherapy (CT) as 1st-line treatment for patients with metastatic colorectal (mCRC): results according to KRAS and BRAF mutations. ECCO-ESMO A6077. Eur J Cancer Supp, 7: 345
Yen LC, Yeh YS, Chen CW, Wang HM, Tsai HL, Lu CY, Chang YT, Chu KS, Lin SR, Wang JY (2009) Detection of KRAS oncogene in peripheral blood as a predictor of the response to cetuximab plus chemotherapy in patients with metastatic colorectal cancer. Clin Cancer Res 15: 4508–4513
Zhang W, Gordon M, Schultheis AM, Yang DY, Nagashima F, Azuma M, Chang HM, Borucka E, Lurje G, Sherrod AE, Iqbal S, Groshen S, Lenz HJ (2007) FCGR2A and FCGR3A polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J Clin Oncol 25: 3712–3718
Acknowledgements
We are grateful to Pr Mario Tosi and Nikky Sabourin for the critical review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Di Fiore, F., Sesboüé, R., Michel, P. et al. Molecular determinants of anti-EGFR sensitivity and resistance in metastatic colorectal cancer. Br J Cancer 103, 1765–1772 (2010). https://doi.org/10.1038/sj.bjc.6606008
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bjc.6606008
Keywords
This article is cited by
-
Combination of KIR2DS4 and FcγRIIa polymorphisms predicts the response to cetuximab in KRAS mutant metastatic colorectal cancer
Scientific Reports (2019)
-
PIK3CA hotspot mutations differentially impact responses to MET targeting in MET-driven and non-driven preclinical cancer models
Molecular Cancer (2017)
-
A synonymous EGFR polymorphism predicting responsiveness to anti-EGFR therapy in metastatic colorectal cancer patients
Tumor Biology (2016)
-
Prediction of response to anti-EGFR antibody-based therapies by multigene sequencing in colorectal cancer patients
BMC Cancer (2015)
-
RasGRP1 opposes proliferative EGFR–SOS1–Ras signals and restricts intestinal epithelial cell growth
Nature Cell Biology (2015)
