
Abstract
Cholestatic liver diseases, accompanied by the hepatic accumulation of bile salts, frequently lead to liver fibrosis, while underlying profibrogenic mechanisms remain incompletely understood. Here, we evaluated the role of extracellular pH (pHe) on bile salt entry and hepatic stellate cell (HSC) activation and proliferation. As modulators of intracellular pH (pHi), various proton pump inhibitors (PPI) were tested for their ability to prevent bile salt entry and HSC activation. Lastly, the PPI pantoprazole was employed in the 3,5-Diethoxycarbonyl-1,4-Dihydrocollidine (DDC)-diet model of cholestatic liver fibrosis. We found in vitro, that slightly acidic pHe (7.2–7.3) enhanced bile salt accumulation in HSC and was a prerequisite to bile salt-induced HSC activation. Pantoprazole in the DDC model exhibited antifibrotic effects. We conclude that bile salt-induced activation of HSC may depend on the slightly acidic microenvironment present in the perisinusoidal space and modulation of pHi in HSC may offer a novel pharmacological target in cholestatic disease.
Similar content being viewed by others


Introduction
Chronic cholestatic liver diseases such as primary biliary cholangitis (PBC), primary sclerosing cholangitis (PSC), and inborn cholestatic syndromes, e.g., progressive familial intrahepatic cholestasis (PFIC), frequently result in liver fibrosis and may progress to liver cirrhosis, ultimately necessitating for liver transplantation1. Despite their varying pathogenetic backgrounds, cholestatic liver diseases share the systemic and hepatic bile accumulation of bile salts2,3. This accumulation of bile salts has been implicated in fibrogenesis in cholestasis, a hypothesis that has been widely accepted since the 1970s2,4. However, mechanisms underlying bile salt-induced fibrogenesis remain scarcely characterized.
Activated hepatic stellate cells (HSCs) represent the main source of extracellular matrix deposition characterizing liver fibrosis. Their activation in cholestatic disease has been attributed, in part, to pro-fibrotic signals such as TGFβ, apoptotic bodies or DNA derived from hepatocytes undergoing bile salt-induced cell death5,6,7,8. However, some of these results have hardly been reproduced since initial discovery and in vivo evidence for the contribution of these mechanisms to liver fibrosis remains scarce.
We have recently shown chenodeoxycholate (CDC), the predominating bile salt accumulating in chronic cholestasis, to induce expansion of cell mass and excess extracellular matrix deposition by human and murine HSCs9,10. This was in line with earlier research indicating activation of pro-proliferative signaling by bile salts11,12,13. Subsequently, we could demonstrate that presence of (G)CDC is crucial for development of liver fibrosis in an in vivo model of hepatocellular cholestasis9. In line with previous reports in bile salt export pump (BSEP) knockout animals14, intrahepatic cholestasis in the absence of (G)CDC, however, did not lead to liver fibrosis9, highlighting the importance of human hydrophobic bile salts for cholestatic liver fibrosis. Bile salt-induced activation of HSCs could subsequently be attributed in vitro to Erk/MEK and PI3K p110α signaling9,10.
In our previous studies, it seemed that only unconjugated CDC but not glycine-conjugated CDC (GCDC), could directly induce proliferation and collagen deposition by HSCs9,10. What are potential explanations for this observation? Activation of signaling pathways by bile salts greatly depends on bile salt uptake and intracellular accumulation15. While sodium taurocholate co-transporting polypeptide (NTCP) is the major bile salt uptake transport in hepatocytes16, it is hardly expressed in dormant HSCs17, and its physiologic role in HSCs bile salt homeostasis has been questioned18. Hydrophobic bile salts can, however, passively cross cell membranes and enter cells. As previously shown for cholangiocytes, passive entry of bile salts is determined by their glycine- or taurine-conjugation or absence thereof and the resulting pKa value of the bile salt (∼4.2 for GCDC compared to ∼4.5 for CDC) as well as by pHe19,20. As a result, glycin-conjugated bile salts require a more acidic pH to become protonated and passively enter living cells.
It is important to consider in this context, that HSCs reside in a compartment of slightly acidic pH21, the perisinusoidal space (or space of Disse). The pHe may thus be a component of the cellular microenvironment crucial for bile salt-induced signaling in HSC.
In the present study we have tested this hypothesis and were able to show that pHe crucially modulates activation of HSC by bile salts and that intracellular pH (pHi) in HSC can be targeted by proton pump inhibitors (PPIs) to prevent such activation. In vivo, we found that the proton pump inhibitor pantoprazole (PPZ) ameliorated liver fibrosis in a mouse model of cholestatic liver fibrosis, indicating that modification of pHi in HSC by targeted therapies might be therapeutically employed.
Results
CDC, but not GCDC, induces activation of HSC under standard cell culture conditions
We have previously shown that presence of GCDC, the predominant hydrophobic bile salt accumulating in chronic cholestatic liver disease in humans, is a prerequisite for the development of liver fibrosis in models of hepatocellular cholestasis9. Here, we tested the ability of GCDC to directly activate HSC in vitro. While CDC in low micromolar concentrations (20–50 µM), expectedly, induced αSMA expression in LX2 (Fig. 1A), GCDC was unable to activate LX2 cells when applied in standard culture medium in concentrations up to 500 µM (Fig. 1B). In light of the considerable cytotoxicity of GCDC at concentrations from 250 µM (Fig. 1C), we did not test stimulatory effects of higher doses. CDC, at the stimulatory concentrations applied, did not induce relevant cytotoxicity (not shown). Bile salt uptake, i.e., intracellular accumulation, was tested by use of the bile salt analog cholyl-lysyl-fluorescein (CLF). We found that CLF after stimulation for 1 h accumulated in LX2, and intracellular fluorescence was dose-dependent (Fig. 1D).

LX2 cells were stimulated with CDC (A) or GCDC (B) for 24 h at the concentrations indicated, and αSMA expression was quantified by western blotting in relation to GAPDH. TGFβ was used as a positive control. C Cell viability was assessed using WST-1 assays after 24 h of stimulation. D LX2 cells were incubated with CLF at pHe 7.3 for 1 h at the indicated concentrations, and intracellular fluorescence was quantified (n = 4–5; p < 0.05, **p < 0.01, compared to control; #p < 0.05, ##p < 0.01, compared to (G)CDC groups; t-test, ANOVA, post hoc LSD or Tukey).
In brief, GCDC, unlike CDC, seems unable to activate HSC at standard cell culture conditions. A possible explanation for this difference is the lower pKa and reduced ability for passive cell entry of GCDC compared to CDC.
Activation of HSC by (G)CDC is critically determined by pHe
In consequence of its lower pKa, GCDC compared to CDC requires a more acidic pH for protonation and passive cell entry. Considering the slightly acidic pH (pH 7.2–7.3) in the space of Disse, where dormant HSCs reside, this might be physiologically relevant21.
Standard culture conditions for LX2 cells have repeatedly been reported as follows: DMEM containing 2% FBS, cultivated in a humidified atmosphere with 5% CO2 and 21% O2 at 37 °C10,22,23,24,25. After allowing this standard culture medium (cell-free) to adjust in the CO2 atmosphere for 24 h, pH was measured and a pH of 7.64 ± 0.1 (n = 5) was found. Adjusting the bicarbonate buffer system in the cell culture medium, pH-controlled medium of pH 7.6, 7.5, 7.4, 7.3 and 7.2 was applied for subsequent experiments. While pH modification was overall well-tolerated, cell viability started to decline from pH 7.2 (not shown).
While GCDC (100 µM) was unable to activate LX2 in a standard culture condition, a 1.9 ± 0.4-fold protein expression of αSMA was induced upon GCDC treatment at pH 7.3 (n = 4, p < 0.01, ANOVA, Tukey) (Fig. 2A). Alteration of extracellular pH alone, had no effect on αSMA protein expression (Supplementary Fig. 1). Picogreen assay was performed to quantify DNA amount as a surrogate for cell proliferation. We found GCDC at 100 µM to induce LX2 proliferation at pHe 7.3, while no stimulation was documented at pH 7.6 (Fig. 2B). According to our hypothesis, increased activation of LX2 by GCDC at lower pH would be a result of increased bile salt uptake. To assess whether bile salt uptake in LX2 is indeed influenced by pHe, we fluorometrically quantified intracellular accumulation of the bile salt analog CLF. As predicted, CLF accumulation increased with stepwise reduction of pHe from 7.6 to 7.3 (Fig. 2C). If the increase in bile salt-induced activation of LX2 at lower pH was a general principle based on a pH-dependent increase in bile salt uptake, the same effect should be detectable for CDC. We therefore applied a lower dose of CDC (10 µM), which had earlier been found to be unable to activate LX2 cells at standard culture conditions, and tested its ability to activate LX2 at various pHe conditions. As seen for GCDC, cells were increasingly activated with lowering pHe. αSMA expression was increased 1.1 ± 0.1, 1.3 ± 0.2 and 1.5 ± 0.3-fold at pH 7.5, 7.4 and 7.3, respectively, compared to control (Fig. 2D). CDC-induced cell proliferation, too, was dependent on pHe in LX2 cells (Fig. 2E).


LX2 cells were stimulated with GCDC (100 µM) for 24 h in buffered culture medium to test various pHe values (7.6–7.2). Subsequently, αSMA protein expression was determined by Western Blotting, and representative images for αSMA protein are shown as well as quantitative analysis of αSMA normalized for GAPDH (A). Furthermore, DNA amount as a surrogate of proliferation was quantified by Picogreen assays (B). LX2 cells were incubated in presence of CLF at various pHe (7.6–7.2) for 1 h, and accumulation was determined fluorometrically (C). LX2 cells were stimulated with CDC (10 µM) for 24 h in buffered culture medium to test various pHe values (7.6–7.2). Subsequently, αSMA protein expression was determined by Western Blotting, and representative images for αSMA protein are shown as well as quantitative analysis of αSMA normalized for GAPDH (D). Furthermore, DNA amount as a surrogate of proliferation was quantified by Picogreen assays (E). After 24 h stimulation with sulfasalazine (10 µM) and myrcludex B (50 nM), LX2 were incubated with CLF for 1 h to determine bile salt accumulation. Amiloride was used as positive control, as will become evident in Fig. 4 (F). αSMA protein expression was determined by Western Blotting, and representative images are shown as well as quantitative analysis of αSMA, normalized for GAPDH (G, H). (n = 4–5; *p < 0.05, **p < 0.01, compared to control; #p < 0.05, ##p < 0.01, compared to (G)CDC groups; t-test, ANOVA, post hoc LSD or Tukey).
The role of active bile salt uptake via specific transporters in HSC is under debate17,18. To exclude transporter-mediated uptake in our system, as opposed to the assumed pH-dependent passive entry, inhibitors of NTCP and OATP were applied, myrcludex B and sulfasalazine, respectively26,27. We found that both inhibitors, sulfasalazine and myrcludex B, neither altered bile salt, i.e., CLF accumulation nor CDC- or GCDC-induced activation for LX2 (Fig. 2F–H).
Taken together, these results suggest that extracellular pH, known to be slightly acidic in the natural microenvironment of HSCs, crucially determines (passive) bile salt uptake and (G)CDC-induced activation and proliferation of HSC.
Pantoprazole leads to intracellular acidification in HSCs and prevents bile salt uptake and bile salt-induced activation of HSCs, partly mediated by inhibition of the sodium‐hydrogen exchanger
While extracellular pH can hardly be pharmacologically addressed, the transcellular pH gradient can be targeted by the alteration of intracellular pH. This might offer a pharmacological target to prevent profibrogenic signaling induced by bile salts. PPZ, the most widely used proton pump inhibitor, is known for inactivating H+/K+-ATPase in the gastric mucosa but may also target alternative proton pumps. In HSC, NHE has previously been implicated in pHi regulation28,29,30. We measured NHE activity in LX2 cells, and found it to be dose-dependently reduced following incubation with PPZ (Fig. 3A). Subsequently, we determined pHi in LX2 following incubation with PPZ and again found a dose-dependent decrease (5–80 µM) (Fig. 3B). Next, to assess whether PPZ can affect bile salt accumulation, CLF was used and was observed to accumulate less as LX2 were pre-treated with PPZ for 24 h (Fig. 3C). To determine whether PPZ can alter pHi in LX2 also in the presence of bile salts, LX2 were simultaneously stimulated with PPZ and GCDC for 24 h. Following incubation with GCDC, LX2 cells were found to have a higher pHi compared to control-treated cells (Fig. 3D). This was in line with previous reports showing that activated HSCs had higher pHi in comparison to dormant HSCs28. Of note, also in the presence of GCDC, PPZ reduced pHi in a dose-dependent manner (Fig. 3D). Decreased pHi and bile salt uptake in the presence of PPZ was associated with an amelioration of GCDC-induced activation of LX2 cells as indicated by a reduction in αSMA and collagen type I alpha I (col1α1) expression (Fig. 3E). Furthermore, proliferation induced by GCDC was prevented by PPZ in a dose-dependent manner, as suggested by Picogreen assays (Fig. 3F). Importantly, WST-1 assays did not show evidence of cell toxicity exerted by PPZ plus GCDC (Fig. 3G). All effects of PPZ on pHi, LX2 activation and proliferation could be reproduced when CDC (20 µM) was applied instead of GCDC (Supplementary Fig. 2). Some effect of PPZ on spontaneous αSMA protein expression in the absence of bile salts in LX2 was observed (Supplementary Fig. 3), indicative of an additional, pleiotropic antifibrotic effect, as reported earlier31.

LX2 cells were pre-treated with PPZ (2.5–80 µM) for 24 h in buffered culture medium at pHe 7.3. Subsequently, Na+/H+ activity (A), pHi (B), and bile salt accumulation (C) were determined. Furthermore, LX2 cells were simultaneously stimulated with GCDC (100 µM) and PPZ (5–80 µM) for 24 h in buffered culture medium at pHe 7.3. Subsequently, pHi was determined (D), as well as αSMA and col1α1 protein expression (E, representative blot and densitometric analysis), DNA amount as a surrogate of proliferation (F) and cell viability, quantified by WST-1 assays (G). (n = 4–6; *p < 0.05, **p < 0.01, compared to control; #p < 0.05, ##p < 0.01, compared to (G)CDC groups; t-test, ANOVA, post hoc LSD or Tukey).
Notably, TGFβ was reported to induce and activate NHE32, leading to a lower pHi. We therefore determined CLF accumulation TGFβ- treated cells compared to controls. We found that CLF accumulation was higher upon TGFβ treatment compared to control (13 ± 1% vs. 4 ± 3% positive cells) (Supplementary Fig. 4). This observation supports our view that higher pHi is associated with an increase in bile salts entrapment in HSCs.
In summary, our results indicate that pHi critically determines bile salt accumulation and bile salt-induced HSC activation and that inhibition of intracellular proton pumps, e.g., the NHE can decrease pHi, prevent bile salt accumulation and inhibit bile salt-induced HSC activation.
(G)CDC-induced HSC activation is ameliorated by PPIs targeting NHE and vacuolar (v)-H+-ATPase
Was the protective effect of PPZ attributable to pHi change or could it be due to pleiotropic effects of PPZ? To further validate a specific, pHi-mediated effect, an additional, established inhibitor of NHE was used, amiloride. Since v-H+-ATPase has been established as an additional potent pHi regulator in HSCs33, its inhibitor bafilomycin A1 was additionally tested. We found that amiloride (10 µM, 100 µM) and bafilomycin A1 (1 nM, 10 nM) were effective in lowering pHi in LX2 in absence (Fig. 4A) and presence (Fig. 4B) of GCDC, and inhibited bile salt accumulation in LX2 (Fig. 4C). In consequence, both amiloride and bafilomycin A1 ameliorated GCDC-induced activation of LX2 as determined by αSMA and col1α1 at protein expression (Fig. 4D) as well as proliferation as determined by Picogreen assays (Fig. 4E). Treatment with PPI alone, i.e., amiloride, notably had no effect on αSMA protein expression at 10 μM (Supplementary Fig. 5). WST-1 assays showed no toxicity of the inhibitors used in presence of GCDC (Fig. 4F). Identical effects were found when CDC was applied instead of GCDC (Supplementary Fig. 6).


LX2 cells were stimulated with amiloride (10 and 100 µM) or bafilomycin A1 (1 and 10 nM) for 24 h in buffered culture medium at pHe 7.3. Based on results from Figs. 2 and 3, PPZ (80 µM) was used as a positive control for pHi alteration. Subsequently, pHi values (A) and bile salt accumulation (B) were determined. Subsequently, experiments were repeated in presence of GCDC (100 µM) and pHi was determined (C). αSMA and col1α1 protein expression were determined by Western Blotting and representative images are shown as well as quantitative analysis of αSMA and col1α1, normalized for GAPDH (D). Furthermore, DNA amount as a surrogate of proliferation was quantified by Picogreen assays (E). Cell viability was quantified by WST-1 assays (F). In independent experiments, LX2 cells were stimulated with TGFβ (10 ng/ml) for 24 h in presence or absence of PPZ (80 µM), amiloride (10 and 100 µM) and bafilomycin A1 (1 and 10 nM), respectively. αSMA protein expression was determined by Western Blotting and representative images are shown as well as quantitative analysis of αSMA, normalized for GAPDH (G). (n = 4–5; *p < 0.05, **p < 0.01, compared to control; #p < 0.05, ##p < 0.01, compared to (G)CDC groups or TGFβ; t-test, ANOVA, post hoc LSD or Tukey).
To further clarify the role of potential pleiotropic, antifibrotic effects of the inhibitors applied, as opposed to specific action via pHi alteration, the effects of PPZ, amiloride and bafilomycin A1 on TGFβ-induced LX2 activation were determined. We found that amiloride 10 and 100 µM did not affect TGFβ-induced LX2 activation (Fig. 4G). PPZ ameliorated TGFβ-induced activation to some extent, although with lower efficacy compared to that seen in bile salt-induced LX2 activation. Bafilomycin A1, however, ameliorated LX2 activation also in this setting, hinting toward an additional pleiotropic effect.
Thus, pHi modification in LX2 by various PPIs may prevent bile salt-induced HSCs activation and proliferation.
GCDC-induced activation of primary murine HSCs is critically determined by pHe and ameliorated by inhibition of intracellular proton pumps
To confirm our results on pH- and proton pump-dependent bile salt-induced activation of LX2, we expanded experiments to primary murine HSCs. While GCDC (100 µM) was unable to activate mHSC in a standard culture condition, a 2.0 ± 0.1-fold protein expression of αSMA was induced at pH 7.2 (Fig. 5A). Next, we assessed whether bile salt uptake in mHSC is influenced by pHe. Similarly to our results in LX2, CLF accumulation increased with stepwise reduction of pHe from 7.6 to 7.2 (Fig. 5B). In line with our experiments in LX2, PPIs were applied in mHSC and we found a decrease in both bile salt accumulation (Fig. 5C) and GCDC-induced mHSC activation (Fig. 5D), while PPI alone had no effect on spontaneous activation (Supplementary Fig. 7). Notably, in previous reports33, activated HSCs were found to present a higher pHi compared to quiescent HSCs. This would predict an increase in bile salt accumulation. In line with this assumption, we found activated mHSCs to have a higher accumulation of CLF compared to quiescent cells (Supplementary Fig. 8).

Primary murine cells were stimulated with GCDC (100 µM) for 24 h after 1 day isolation in buffered culture medium to test various pHe values (7.6–7.2). Subsequently, αSMA protein expression was determined by Western Blotting and representative images are shown as well as quantitative analysis of αSMA, normalized for GAPDH (A). mHSC cells were incubated in presence of CLF at various pHe (7.6–7.2) for 1 h and accumulation was determined fluorometrically (B). mHSC cells were stimulated with PPZ (80 µM), amiloride (10 and 100 µM) or bafilomycin A1 (1 and 10 nM) for 24 h in buffered culture medium at pHe 7.2, bile salt accumulation (C) was determined. Subsequently, experiments were repeated in presence of GCDC (100 µM). αSMA protein expression was determined by Western Blotting and representative images are shown as well as quantitative analysis of αSMA, normalized for GAPDH (D). (n = 3–5; *p < 0.05, **p < 0.01, compared to control; #p < 0.05, ##p < 0.01, compared to (G)CDC groups; t-test, ANOVA, post hoc LSD or Tukey).
In brief, an acidic (extracellular) microenvironment appears to be a prerequisite for bile salt-induced activation of primary murine HSCs. In line with our data in LX2 cells, inhibition of intracellular bile salt accumulation by use of various PPIs was able to prevent bile salt-induced activation of mHSCs.
Pantoprazole prevents liver fibrosis in the DDC-diet model of chronic cholestasis
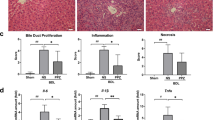
Since proton pump inhibition in HSC prevented bile salt-induced fibrogenesis in vitro, we aimed at translating our results into an in vivo system. Among available PPIs, PPZ is the clinically best established. Therefore, to test its potential to prevent liver fibrosis in cholestasis in vivo, PPZ was applied in the DDC-diet model. Evaluation of H&E stains showed minor tissue damage but significant inflammation in DDC-fed mice, which was unaltered upon PPZ administration (Fig. 6A, B). Despite a lack in improvement of liver inflammation, Sirius red, as well as Masson staining, detected a decrease in DDC-induced liver fibrosis following PPZ treatment (Fig. 6A, C, D). This was confirmed by immunohistochemistry for αSMA (Fig. 6A, E). In serum biochemistry, ALT and AST as markers for liver damage as well as alkaline phosphatase as a marker of cholestasis were markedly increased in DDC-fed mice, as expected. No improvement in these biochemical markers was observed when PPZ was applied (Fig. 6F–H), indicating that the severity of primary liver damage was not altered by PPZ. Total bilirubin, however, representing a serum marker of advanced liver disease, i.e., fibrosis, was slightly but significantly improved upon PPZ administration (Fig. 6I). Furthermore, pro-fibrotic markers TIMP1 and PDGFB were significantly downregulated by PPZ on mRNA level (Fig. 6J, K).

C57BL/6 male mice aged 4–6 weeks were fed with control or DDC diet (0.1%) and were administered with H2O as control or PPZ at 5 mg/kg, twice a day, i.p., for 4 weeks. Representative images for general macroscopic appearance, H&E, Masson staining, Sirius red staining and IHC for αSMA are given in (A), scale bar:100 μm. Liver inflammation was semi-quantitatively assessed (B) and quantitative assessment (% of total area) is given for Masson staining (C), Sirius red staining (D) and IHC for αSMA (E) is presented. From serum biochemistry, levels of ALT (F), AST (G), alkaline phosphatase (H) and total bilirubin (I) are given. For rt-qPCR, mRNA expressions of pro-fibrotic markers, TIMP1 (J) and PDGFB (K), are reported. (n = 5–7; *p < 0.05; **p < 0.01, compared to control; #p < 0.05; ##p < 0.01, compared to DDC + H2O; t-test, ANOVA, post hoc LSD or Tukey).
Taken together, these in vivo data suggest that PPZ does not prevent cholestasis, liver damage or liver inflammation in the DDC mouse model of chronic cholestasis but specifically ameliorates activation of profibrogenic pathways and liver fibrosis.
Discussion
Accumulation of toxic bile salts in chronic cholestatic liver disease is believed to induce and promote liver fibrosis. However, underlying mechanisms for bile salt-induced fibrogenesis remain incompletely characterized. We have previously shown direct induction of proliferation and extracellular matrix deposition by bile salts in HSC9,10. We had observed, however, that GCDC, the predominant bile salt accumulating in chronic cholestatic liver disease in man, was less effective in activating HSCs compared to unconjugated CDC. This led us to reconsider the microenvironment in which HSCs reside, the slightly acidic perisinusoidal space, as pH has been shown to be an important co-factor for bile salt-induced signaling in cholangiocytes19.
In the current study, we found that GCDC, unlike CDC, was unable to activate LX2 cells in standard culture conditions (Fig. 1). Subsequently, we could demonstrate that a slightly acidic extracellular pH (pH 7.3) was a prerequisite for GCDC-induced activation of LX2 (Fig. 2). Acidic pHe led to enhanced intracellular bile salt accumulation and GCDC-induced αSMA expression and proliferation. Given the microenvironment of a slightly acidic pH in the perisinusoidal space, this observation is of potential pathophysiologic relevance. Since pHe in the portal bloodstream can hardly be targeted therapeutically, we investigated the effects of modifying intracellular pH in HSC. PPZ as a model PPI was indeed able to lower intracellular pH in LX2 cells, and we could attribute this effect to inhibition of NHE (Fig. 3A). Lower pHi in LX2 was associated with a reduction in bile salt accumulation and inhibited bile salt-induced αSMA- and collagen1α1-expression as well as bile salt-induced proliferation (Fig. 3 and Supplementary Fig. 2). These observations could be reproduced when alternative PPIs were used, namely the NHE inhibitor amiloride and the vacuolar-ATPase inhibitor bafilomycin A1 (Figs. 4, 5 and Supplementary Fig. 6). Subsequently, we could confirm our findings in an independent experimental model, by use of primary murine HSCs. In this system, too, GCDC-induced activation was pHe-dependent and could be ameliorated by application of PPIs (Fig. 5). Inversely, we found that TGFβ, which was reported to induce and activate NHE32, increased CLF accumulation in LX2 cells (Supplementary Fig. 3). Furthermore, activated HSCs, which are known to develop an increased pHi compared to quiescent HSCs showed an increase in CLF accumulation compared to quiescent HSCs.
Lastly, PPZ was tested in a mouse model of chronic cholestatic liver fibrosis. PPZ did not alter cholestasis, liver damage or inflammation in the DDC-mouse model but ameliorated liver fibrosis, indicative of a specific antifibrotic effect of PPZ (Fig. 6).
We conclude from our results that passive entry of bile salts in the form of protonated, apolar bile acids into HSCs may be critically influenced by pHe and bile acid pKa, the latter being dependent on bile salt conjugation (pKa ∼4.2 for GCDC compared to ∼4.5 for CDC). This is in line with previous studies in cholangiocytes19. We furthermore conclude that pharmacologically addressing pHi in HSC may serve to prevent bile salt accumulation and HSC activation in chronic cholestasis. How would lowering the pHi influence intracellular bile salt accumulation? Upon passive entry of a protonated bile acid into the cell, the acid would be deprotonated to the corresponding bile salt due to the more alkaline pH present in the intracellular compartment. The bile salt, carrying an electronic charge, would then be unable to passively exit the cell and would be ‘trapped’ intracellularly. Over time, this would lead to accumulation of bile salts within the cells. We acknowledge that measuring CLF accumulation in HSCs rather than influx and efflux of bile acids in our study is an important limitation. However, our findings and conclusions are in line with a very recent report by Kersten et al., who demonstrated that lowering pHi (by incubation with Roachlamide) was able to decrease 3H-GCDC influx in another liver cell type, namely cholangiocytes34. Our results seem in contrast with Svegliati et al. and Sommerfeld et al.11,12, who reported pro-proliferative effects of bile salts in rat HSC even at standard culturing conditions. However, little consideration had then been given to the importance of the pH microenvironment and authors did not report the pH of their culturing condition. Thus, we cannot exclude that these may have been more acidic than our standard culture conditions. Importantly, both before mentioned studies were performed in rat HSC, while our experiments were performed in mouse and human HSCs, respectively, which may also account for the different findings.
It is well established that bile salts as hydrophobic substances can passively enter cells as bile acids, i.e., when being protonated and apolar. In the presence of an active bile salt uptake system, however, passive entry would most likely be of minor relevance. NTCP, as the major bile salt uptake transporter in hepatocytes, has been speculated to be present in HSCs17, but recent evidence suggests that its expression might not be associated with relevant bile salt uptake18. To further exclude a relevant contribution of active bile salt uptake to bile salt-induced activation in LX2, we applied inhibitors for NTCP and OATPs, myrcludex B and sulfasalazine, respectively. We found that inhibition of both bile salt uptake transporters was without effect on (G)CDC-induced LX2 activation, suggesting a predominant role for passive bile acid entry in our system.
Another means of bile salt-induced LX2 activation, independent of passive entry, would be the activation of membrane receptors. The best-characterized bile acid receptor on HSCs, however, TGR5, is primarily expressed in activated HSCs, but not silent HSCs35,36. Furthermore, TGR5 has a very low affinity for (G)CDC35. Thus, the role of TGR5 for (G)CDC-induced activation of dormant HSC seems negligible.
In this study, we found PPZ to lower intracellular pH in LX2 cells, likely by inhibition of NHE. This was associated with a decrease in intracellular bile salt accumulation and subsequent activation of LX2. PPIs are known, however, to have effects beyond proton pump inhibition, e.g., anti-inflammatory and anti-oxidative properties37,38. Thus, these pleiotropic effects of PPZ may have contributed to the observed effects on HSC activation. To exclude a solely antifibrotic class effect of PPIs, we tested additional proton pump inhibitors, chemically different from PPZ. Both the NHE inhibitor amiloride and the v-H+-ATPase inhibitor bafilomycin A1 were demonstrated to also lower pHi. This, too, was associated with reduced bile salt accumulation and activation of LX2. This supports our view, that proton pump-inhibition prevents LX2 activation specifically by pHi alteration and inhibition of intracellular bile salt accumulation. Furthermore, we tested the effect of PPZ, amiloride and bafilomycin A1 on TGFβ-induced LX2 activation. While bafilomycin A1 indeed was antifibrotic in this setting, PPZ was less effective in lowering TGFβ-induced LX2 activation than in lowering bile salt-induced LX2 activation and amiloride was without antifibrotic properties upon TGFβ-stimulation. Thus, while we cannot completely exclude additional, pleiotropic effects of PPZ and other PPIs, part of their antifibrotic effects in our systems seem indeed to be attributable to pHi lowering and prevention of bile salt entry.
To translate our findings into an in vivo system, the DDC-diet mouse model was applied. The dosage of PPZ applied in humans ranges from approximately 0.66 mg/kg body weight in long-term care (40 mg qd for a 60 kg person) up to 4 mg/kg in acute upper GI bleeding (10 mg per hour for a 60 kg person). Given the increased metabolism in mice, translational mouse models are usually applied with higher doses compared to humans. Thus, the 5 mg/kg body weight applied here is well within a standard therapeutic range. Our report is the first one to indicate a therapeutic effect of PPZ on liver fibrosis in mice. Noteworthy, PPZ had little or no effect on the cholestatic phenotype, liver inflammation or liver cell damage, but specifically prevented HSC expansion (as determined by IHC for αSMA) and liver fibrosis. Our results are in line with a recent report by Lu et al., who demonstrated an antifibrotic effect of PPZ in bile duct ligation rats31. They attributed this finding to increased YAP degradation, while our results suggest an additional mode of action via pHi alteration in HSC. However, due to high dosage of PPZ used in vitro (100–300 μM) and in vivo (150 mg/kg)31, these results need to be interpreted with caution. In fact, these doses had previously been associated with profibrogenic rather than therapeutic effects, when Assalin et al. found that PPZ induced liver fibrosis in mice (150 mg/kg for 60 days)39.
In patients with liver cirrhosis, PPI intake has recently been associated with an increased probability of decompensation in a retrospective cohort40. This possibly can be explained by an increased sensibility toward infections in this highly susceptible cohort and does not necessarily exclude beneficial effects on fibrogenesis when PPI are applied in early stages of disease. Furthermore, our results suggest that other pHi modulating agents for specific uptake by HSCs might deserve further development, which might come without the increased risk of infection that is associated with PPZ administration.
Our results suggest that passive, pHe-mediated bile salt entry into HSCs triggers their activation, highlighting the role of the acidic microenvironment of HSCs in the perisinusoidal space (Fig. 7). Both bile salt entry and activation of HSC can be prevented via pHi modulation by PPIs (Fig. 7). This provides a novel pharmacological target for antifibrotic therapies in chronic cholestatic liver disease, meriting further exploration.

In cholestatic liver disease, the relatively acidic pHe in the perisinusoidal space favors the passive entry of protonated bile salt and the relatively alkaline pHi leads to ionized bile salt accumulation in HSCs, which subsequently induce HSCs activation. Upon pHi modification treatment, bile salt-induced HSCs activation is inhibited by preventing bile salt accumulation via acidifying pHi.
Materials and methods
Primary murine hepatic stellate cells and cell culture
Primary murine HSCs were isolated from male C57BL/6 mice at the age of 6 to 12 weeks and purified by pronase-collagenase perfusion followed by density gradient centrifugation in 13.2% Nycodenz (Axis-Shield PoC, Oslo, Norway), as described previously41. LX2 cells were purchased from Merck-Millipore, Darmstadt, Germany. Cells were cultured in high glucose (LX2) or low glucose (mHSC) Dulbecco’s Modified Eagle Medium (DMEM) (Sigma-Aldrich, Taufkirchen, Germany) containing 2% (LX2) fetal bovine serum (FBS, PAN-Biotech, Aidenbach, Germany) and 1% antibiotics (Sigma-Aldrich, Germany) as described previously10,22,23,24,25. Cell medium at pH of 7.6–7.2 was generated by adjusting the bicarbonate buffer system through proportionately mixing of regular DMEM and DMEM without sodium bicarbonate. Cells were cultivated in a humidified atmosphere with 5% CO2 and 21% O2 at 37 °C. LX2 cells were stimulated with TGF-β (Peprotech, Germany), chenodeoxycholate (CDC, Sigma-Aldrich, Taufkirchen, Germany), glycochenodeoxycholate (GCDC Sigma-Aldrich, Darmstadt, Germany), pantoprazole (PPZ), bafilomycin A1 (CST, Germany), sulfasalazine (Sigma-Aldrich, Germany) and myrcludex B (Sigma-Aldrich, Germany), respectively at indicated concentrations.
Viability assay
Cell viability was measured by WST assay (Roche, Mannheim, Germany), according to the manufacturer’s instructions, with the EASY READER SFplus (SLT-Labinstruments, Salzburg, Austria).
Intracellular pH measurement
The pHi of cells was measured by using the cell-permeable probe 3′-O-acetyl-2′,7′-bis(carboxyethyl)-5,6-carboxyfluoresceinacetoxymethylester (BCECF-AM). Cells were cultured in a 96-well plate at 37 °C and then incubated with BCECF-AM for 30 min at 37 °C in full humidity with 5% CO2. Cells were washed twice with the HEPES buffer. Fluorescence intensity was determined using a fluorescent plate reader with an excitation wavelength of 450 nm/490 nm, and 535 nm emission wavelength (PerSeptive Biosystems, Framingham, MA, USA). pH calibration was made applying high K+ solutions (140 mM) at different pH values (7.8, 7.4, 7.0, 6.6, 6.0), buffered with HEPES or MOPS and 10 µM Nigericin (Invitrogen, USA).
Measurement of bile salt accumulation in LX2
Bile salt accumulation in LX2 was evaluated by fluorometric quantification of Cholyl-Lysyl-Fluorescein (CLF), a bile salt analog. Following pre-incubation at indicated pHe and in presence or absence of indicated inhibitors, cells were incubated with CLF at indicated concentrations at 37 °C for 30 min and washed twice with HBSS. Fluorescence measurement was performed with 100 µL/well of HEPES buffer in a plate reader (Cyto Fluor 4000 PerSeptive Biosystems, Framingham, MA, USA) at wavelength of excitation 485 nm and emission 530 nm.
DNA quantification
Total DNA amount in cell culture as a surrogate of cell number was determined by the PicoGreen® dsDNA assay (Invitrogen, Carlsbad, USA) according to the manufacturer’s protocol, with a CytoFluor Multi-Well Plate Reader Series 4000 (PerSeptive Biosystems, Framingham, MA, USA).
Reverse-transcription quantitative polymerase chain reaction (RT-qPCR)
mRNA gene expression of PDGFB, TMIP1 and β-ACTIN were measured by RT-qPCR. Total RNA from mouse liver tissue was isolated with TranszolUp reagent (TransGen Biotech, China) according to the manufacturer’s instructions. Purity and concentration of the isolated RNA were determined by the NanoPhotometer N60/N50 (IMPLEN). Total purified RNA was amplified by PerfectStart Green qPCR SuperMix (TransGen Biotech, China) and translated into cDNA by EasyScript® One-Step gDNA Removal and cDNA synthesis SuperMix (TransGen Biotech, China). Quantitative PCR was performed with CFX ConnectTM Real-Time System (Bio-Rad, USA). All qPCR primers used are listed in Table 1. mRNA Expression was calculated according to ΔΔCT method with β-actin as the housekeeping gene and normalized to the means of the controls.
Western blotting
Proteins were loaded in equal amounts, separated by SDS-PAGE and transferred onto PVDF membrane (Merck-Millipore, Darmstadt, Germany). Membranes were blocked in 1% casein (Carl Roth, Karlsruhe, Germany) for 30 min and incubated with primary antibodies against α-smooth muscle actin (αSMA, Sigma-Aldrich, 1:1000; Catalog number: A2547), collagen type I alpha I (col1α1, Abcam, 1:1000; Catalog number: ab6308) and GAPDH (Abcam, 1:5000; Catalog number: ab8245) overnight at 4 °C, followed by incubation with secondary goat anti-mouse IgG-HRP (Bio-Rad, 1:20,000; Catalog number: #170-0616, Feldkirchen, Germany). Visualization was performed with Clarity™ Western ECL Substrate (Super SignalTM West Femto, Thermo Scientific Rockford, USA), detected with the ChemoCam (INTAS, Göttingen, Germany)
Na+/H+ activity measurement
Na+/H+ activity was determined as reported earlier42 by studying pHi change upon removal and re-introduction of extracellular Na+: LX2 cells were seeded on 24*24 mm glass cover slides in φ35 mm petri dish. Cells were divided into control and PPZ (2.5, 5, 10, 20, 40, 80 μM) treatment groups when the cell confluency reached 60–80%. After 24 h of PPZ treatment, cells were washed twice with Na+-containing HEPES buffer. Cells were incubated at 37 °C for 30 min in Na+-containing HEPES with 5 μM BCECF-AM. Subsequently, cells were washed twice. Glass slides were placed on the microscope stage of Ratio Photometry and Imaging Systems (PTI, USA) and continuously perfused with Na+-free HEPES-buffered solutions by gravity (0–3 min: Na+-free HEPES buffer; 3–8 min: Na+-containing HEPES buffer with 10 μM Nigericin; 8–11 min: Na+-free HEPES buffer with 1% BSA; 11–14 min: Na+-free HEPES buffer; 14–20 min: Na+-containing HEPES buffer). BCECF-AM module of PTI was applied to measure pHi of cells with Na+-free HEPES buffer. The fluorescent dye was excited at 440 and 485 nm alternatively and recorded every 30 s. pHi alteration of cells during 14–17 min was calculated to represent Na+/H+ exchanger activity.
Animals
All animal protocols were approved by the Animal Ethics Committee of Zunyi Medical University and performed in accordance with the Good Laboratory Practice regulations for non-clinical laboratory studies of drugs issued by the National Scientific and Technological Committee of the People’s Republic of China. Male C57BL/6J mice (8 weeks of age) were purchased from SPF (Beijing) Biotechnology Co., Ltd., and they were maintained at 22 °C with a 12‐h:12‐h light/dark cycle and had free access to normal chow diet and water.
DDC model
Mice were randomly allocated into three groups: control, DDC-sterile H2O, DDC-PPZ. Mice in the control group were fed with control diet and mice in the DDC groups were fed with 0.1% DDC-containing diet. DDC-diet mice were injected i.p. by sterile H2O or 5 mg/Kg PPZ dissolved in sterile H2O twice per day for 4 weeks. Mice were sacrificed after 4 weeks of treatment. Liver tissue and plasma were collected. We have complied with all relevant ethical regulations for animal use.
Serum analysis and liver histology
Serum aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase (ALP) and total bilirubin (TBIL) were measured by Beckman AU5800 Chemistry Analyzer using kits purchased from Beckman Coulter Inc. (USA). Liver samples were fixed with 4% formaldehyde. After embedding in paraffin, 3 μM sections were stained with hematoxylin and eosin (H&E), Sirius red and Masson staining according to standard protocols. Liver inflammation was quantified via blinded scoring 0–4: 0: No inflammation; 1: Slightly chronic inflammation; 2: Mild chronic inflammation; 3: Mild inflammation; 4: Severe inflammation.
Immunohistochemistry
Paraffin-embedded sections (3 μM) of liver tissues were used for immunohistochemical staining. Anti-α-SMA antibody (αSMA, abcam, 1:1000; Catalog number: ab124964) were applied as primary antibody. Polyperoxidase-anti-Mouse/Rabbit IgG (Elabscience, Ready-to-Use, Catalog number: E-IR-R217B) was applied as second antibody. Specific staining was visualized by light microscopy.
Statistics and reproducibility
All in vitro experiments were repeated at least 3 times. For all animal experiments, at least 5 samples were included in each group. Statistical calculations were performed by using SPSS 25 (IBM, USA) or GraphPad Prism 7 (USA) using analysis of t-test, ANOVA with appropriate post hoc tests (Fisher’s LSD or Tukey’s). P-values lower than 0.05 were considered statistically significant. All data are presented as mean ± standard deviation.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All data supporting the results and conclusions of this paper are available upon request from the corresponding authors. All data behind the graphs and charts shown in the figures are presented in Supplementary Data. Uncropped and unedited blot images are available in Supplementary Information.
Abbreviations
- ALT:
-
Alanine aminotransferase
- AST:
-
Aspartate aminotransferase
- αSMA:
-
α-smooth muscle actin
- ALP:
-
Alkaline phosphatase
- Bafl A1:
-
Bafilomycin A1
- BCECF-AM:
-
3′-O-acetyl-2′,7′-bis(carboxyethyl)-5,6-carboxyfuoresceinacetoxymethylester
- BSEP:
-
Bile salt export pump
- CDC:
-
Chenodeoxycholate
- CLF:
-
Cholyl-Lysyl-Fluorescein
- col1α1:
-
Collagen type I alpha I
- DDC:
-
3,5-Diethoxycarbonyl-1,4-Dihydrocollidine
- DMEM:
-
Dulbecco’s Modified Eagle Medium
- GCDC:
-
Glycochenodeoxycholate
- TGR5:
-
G-protein-coupled receptor
- HSCs:
-
Hepatic stellate cells
- NTCP:
-
Sodium taurocholate co-transporting polypeptide
- PFIC:
-
Progressive familial intrahepatic cholestasis
- PBC:
-
Primary biliary cholangitis
- pHe:
-
Extracellular pH
- pHi:
-
Intracellular pH
- PPZ:
-
Pantoprazole
- PPI:
-
Proton pump inhibitor
- PSC:
-
Primary sclerosing cholangitis
- TBIL:
-
Total bilirubin
- TGF-β:
-
Transforming growth factor-β
- v-H+-ATPase:
-
Vacuolar-ATPase
References
Adam, R. et al. 2018 Annual Report of the European Liver Transplant Registry (ELTR)—50-year evolution of liver transplantation. Transpl. Int. 31, 1293–1317 (2018).
Greim, H. et al. Mechanism of cholestasis. 6. Bile acids in human livers with or without biliary obstruction. Gastroenterology 63, 846–850 (1972).
Greim, H. et al. Mechanism of cholestasis. 5. Bile acids in normal rat livers and in those after bile duct ligation. Gastroenterology 63, 837–845 (1972).
Dyrszka, H., Salen, G., Zaki, F. G., Chen, T. & Mosbach, E. H. Hepatic toxicity in the rhesus monkey treated with chenodeoxycholic acid for 6 months: biochemical and ultrastructural studies. Gastroenterology 70, 93–104 (1976).
Canbay, A. et al. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab. Invest. 83, 655–663 (2003).
Zhan, S. S. et al. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 43, 435–443 (2006).
Watanabe, A. et al. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology 46, 1509–1518 (2007).
Myung, S. J. et al. Bile acid-mediated thrombospondin-1 induction in hepatocytes leads to transforming growth factor-beta-dependent hepatic stellate cell activation. Biochem. Biophys. Res. Commun. 353, 1091–1096 (2007).
Hohenester, S. et al. Glycochenodeoxycholate promotes liver fibrosis in mice with hepatocellular cholestasis. Cells 9, 281 (2020).
Zimny, S. et al. Hydrophobic bile salts induce pro-fibrogenic proliferation of hepatic stellate cells through PI3K p110 alpha signaling. Cells 11, 2344 (2022).
Svegliati-Baroni, G. et al. Bile acids induce hepatic stellate cell proliferation via activation of the epidermal growth factor receptor. Gastroenterology 128, 1042–1055 (2005).
Sommerfeld, A., Reinehr, R. & Häussinger, D. Bile acid-induced epidermal growth factor receptor activation in quiescent rat hepatic stellate cells can trigger both proliferation and apoptosis. J. Biol. Chem. 284, 22173–22183 (2009).
Saga, K. et al. Secondary unconjugated bile acids induce hepatic stellate cell activation. Int. J. Mol. Sci. 19, 3043 (2018).
Wang, R. et al. Targeted inactivation of sister of P-glycoprotein gene (spgp) in mice results in nonprogressive but persistent intrahepatic cholestasis. Proc. Natl Acad. Sci. USA 98, 2011–2016 (2001).
Komichi, D. et al. Unique inhibition of bile salt-induced apoptosis by lecithins and cytoprotective bile salts in immortalized mouse cholangiocytes. Dig. Dis. Sci. 48, 2315–2322 (2003).
Jani, M. et al. Kinetic characterization of bile salt transport by human NTCP (SLC10A1). Toxicol. Vitr. 46, 189–193 (2018).
Salhab, A., Amer, J., Lu, Y. & Safadi, R. Sodium+/taurocholate cotransporting polypeptide as target therapy for liver fibrosis. Gut 71, 1373–1385 (2022).
Kunst, R. F., Paulusma, C. C. & van de Graaf, S. F. J. Insufficient evidence for NTCP activity in stellate cells. Gut 71, 2140–2141 (2021).
Hohenester, S. et al. A biliary HCO3− umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology 55, 173–183 (2012).
Beuers, U. et al. The biliary HCO3− umbrella: a unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology 52, 1489–1496 (2010).
Ichikawa, M. et al. Effect of perfusate pH on the influx of 5-5′-dimethyl-oxazolidine-2,4-dione and dissociation of epidermal growth factor from the cell-surface receptor: the existence of the proton diffusion barrier in the Disse space. J. Hepatol. 20, 190–200 (1994).
Reiter, F. P. et al. p70 ribosomal protein S6 kinase is a checkpoint of human hepatic stellate cell activation and liver fibrosis in mice. Cell Mol. Gastroenterol. Hepatol. 13, 95–112 (2022).
Franko, A. et al. cGMP-dependent protein kinase I (cGKI) modulates human hepatic stellate cell activation. Metabolism 88, 22–30 (2018).
da Silva, C. M. et al. Cellular and molecular effects of Baccharis dracunculifolia D.C. and Plectranthus barbatus Andrews medicinal plant extracts on retinoid metabolism in the human hepatic stellate cell LX-2. BMC Complement. Alter. Med. 19, 222 (2019).
Carson, J. P., Robinson, M. W., Ramm, G. A. & Gobert, G. N. RNA sequencing of LX-2 cells treated with TGF-β1 identifies genes associated with hepatic stellate cell activation. Mol. Biol. Rep. 48, 7677–7688 (2021).
Donkers, J. M., Appelman, M. D. & van de Graaf, S. F. J. Mechanistic insights into the inhibition of NTCP by myrcludex B. JHEP Rep. 1, 278–285 (2019).
Karlgren, M. et al. Classification of inhibitors of hepatic organic anion transporting polypeptides (OATPs): influence of protein expression on drug-drug interactions. J. Med. Chem. 55, 4740–4763 (2012).
Di Sario, A. et al. Intracellular pH regulation and Na+/H+ exchange activity in human hepatic stellate cells: effect of platelet-derived growth factor, insulin-like growth factor 1 and insulin. J. Hepatol. 34, 378–385 (2001).
Di Sario, A. et al. Characterization of ion transport mechanisms regulating intracellular pH in hepatic stellate cells. Am. J. Physiol. 273, G39–G48 (1997).
Benedetti, A. et al. Inhibition of the Na+/H+ exchanger reduces rat hepatic stellate cell activity and liver fibrosis: an in vitro and in vivo study. Gastroenterology 120, 545–556 (2001).
Lu, Z. N. et al. Pantoprazole ameliorates liver fibrosis and suppresses hepatic stellate cell activation in bile duct ligation rats by promoting YAP degradation. Acta Pharm. Sin. 42, 1808–1820 (2021).
Matsuki, K., Hathaway, C. K., Lawrence, M. G., Smithies, O. & Kakoki, M. The role of transforming growth factor β1 in the regulation of blood pressure. Curr. Hypertens. Rev. 10, 223–238 (2014).
Marrone, G. et al. The adenosine monophosphate-activated protein kinase-vacuolar adenosine triphosphatase-pH axis: a key regulator of the profibrogenic phenotype of human hepatic stellate cells. Hepatology 68, 1140–1153 (2018).
Kersten, R. et al. Galectin-3 and prohibitin 1 are autoantigens in IgG4-related cholangitis without clear-cut protective effects against toxic bile acids. Front. Immunol. 14, 1251134 (2023).
Xie, G. et al. Conjugated secondary 12α-hydroxylated bile acids promote liver fibrogenesis. EBioMedicine 66, 103290 (2021).
Keitel, V., Donner, M., Winandy, S., Kubitz, R. & Häussinger, D. Expression and function of the bile acid receptor TGR5 in Kupffer cells. Biochem. Biophys. Res. Commun. 372, 78–84 (2008).
De Milito, A. et al. Proton pump inhibitors induce apoptosis of human B-cell tumors through a caspase-independent mechanism involving reactive oxygen species. Cancer Res. 67, 5408–5417 (2007).
Kedika, R. R., Souza, R. F. & Spechler, S. J. Potential anti-inflammatory effects of proton pump inhibitors: a review and discussion of the clinical implications. Dig. Dis. Sci. 54, 2312–2317 (2009).
Assalin, H. B. et al. Proton pump inhibitor pantoprazole modulates intestinal microbiota and induces TLR4 signaling and fibrosis in mouse liver. Int. J. Mol. Sci. 23, 13766 (2022).
Mahmud, N., Serper, M., Taddei, T. H. & Kaplan, D. E. The association between proton pump inhibitor exposure and key liver-related outcomes in patients with cirrhosis: a veterans affairs cohort study. Gastroenterology 163, 257–269.e256 (2022).
Friedman, S. L. & Roll, F. J. Isolation and culture of hepatic lipocytes, Kupffer cells, and sinusoidal endothelial cells by density gradient centrifugation with Stractan. Anal. Biochem. 161, 207–218 (1987).
Ebata, S. et al. Aldosterone activates Na+/H+ exchange in vascular smooth muscle cells by nongenomic and genomic mechanisms. Kidney Int. 56, 1400–1412 (1999).
Acknowledgements
We are grateful for assistance in scientific writing provided by Dr. Jutta Nagel, LMU Munich. This work was supported by the German Research Council (grant HO 4460/3-1 to S.H.) and National Natural Science Council of China (82073087) to B.T.
Author information
Authors and Affiliations
Contributions
Study concept and design: S.H., J.L., B.T.; acquisition of data: J.L., S.Y., H.J., R.W.; analysis and interpretation of data: J.L., S.H., S.Z., D.K., R.W., G.D., B.T.; drafting of the manuscript: J.L.; critical revision of the manuscript for important intellectual content: S.H., B.T.; statistical analysis: J.L., R.W.; administrative, technical or material support: S.H., R.W., S.Y., H.J.; study supervision: S.H., B.T.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Yuping Chen and the other anonymous reviewers for their contribution to the peer review of this work. Primary handling editor: Christina Karlsson Rosenthal. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, J., Yao, S., Zimny, S. et al. The acidic microenvironment in the perisinusoidal space critically determines bile salt-induced activation of hepatic stellate cells. Commun Biol 7, 1591 (2024). https://doi.org/10.1038/s42003-024-07192-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-07192-4
