
Abstract
Delta-like protein 3 (DLL3) is highly expressed in solid tumors, including neuroendocrine carcinomas/neuroendocrine tumors (NEC/NET). Rovalpituzumab tesirine (Rova-T) is a DLL3-targeting antibody-drug conjugate. Patients with NECs and other advanced DLL3-expressing tumors were enrolled in this phase I/II study (NCT02709889). The primary endpoint was safety. Two hundred patients were enrolled: 101 with NEC/NET (large-cell NEC, gastroenteropancreatic NEC, neuroendocrine prostate cancer, and other NEC/NET) and 99 with other solid tumors (melanoma, medullary thyroid cancer [MTC], glioblastoma, and other). The recommended phase II dose (RP2D) was 0.3 mg/kg every 6 weeks (q6w) for two cycles. At the RP2D, grade 3/4 adverse events included anemia (17%), thrombocytopenia (15%), and elevated aspartate aminotransferase (8%). Responses were confirmed in 15/145 patients (10%) treated at 0.3 mg/kg, including 9/69 patients (13%) with NEC/NET. Rova-T at 0.3 mg/kg q6w had manageable toxicity, with antitumor activity observed in patients with NEC/NET, melanoma, MTC, and glioblastoma.
Similar content being viewed by others
Introduction
Delta-like protein 3 (DLL3) is a ligand in the Notch signaling pathway that is highly expressed in tumors of neuroendocrine origin but not in normal tissues1,2. The Notch signaling pathway regulates cell proliferation, differentiation, and cell death and may have tumor-suppressive or oncogenic effects, depending on the tissue microenvironment3. Suppression of the NOTCH gene has been shown to promote oncogenesis in small cell lung cancer (SCLC), medullary thyroid carcinoma (MTC), and pancreatic and biliary neuroendocrine tumors3,4. Although the function of DLL3 is not fully understood, it has been implicated in the inhibition of the Notch signaling pathway in the regulation of cell development and cell fate decisions3,5.
Neuroendocrine carcinomas (NEC) are a group of poorly differentiated neuroendocrine neoplasms3 that commonly express DLL3, with positive DLL3 expression observed in 65–74% of large cell NEC and in 77% of castration-resistant neuroendocrine prostate cancer (NEPC)6,7. In contrast, DLL3 expression is not observed at a high prevalence in low-grade, well-differentiated neuroendocrine tumors (NET). Due to the heterogeneity and rarity of neuroendocrine neoplasms, they are understudied and poorly understood3. Platinum-based chemotherapy is a standard first-line option for NEC, despite the lack of survival advantage demonstrated in randomized trial8,9,10. Overall survival (OS) in patients with NEC is <18 months9,10,11,12,13. A significant need beyond first-line therapy exists for novel therapeutic treatment options for patients with NEC, and DLL3 is a potential therapeutic target.
In addition to NEC, other cancers have high DLL3 expression, including melanoma, MTC, and glioblastoma (GBM)14. Patients with metastatic melanoma are typically treated with immune checkpoint inhibitors and BRAF and MEK inhibitors, which produce high response rates with impressive durability. However, metastatic melanoma will ultimately become refractory to these therapies, and median progression-free survival (PFS) is <1 year with these agents15,16. MTC makes up 1–2% of thyroid cancers, and 10–15% of patients present with metastatic disease at diagnosis. These patients are typically treated with a multikinase inhibitor, such as cabozantinib or vandetanib, or with selpercatinib or other RET inhibitors for those with RET-mutated tumors17. However, new treatment options are needed for patients with MTC who do not benefit from or are intolerant to these agents. GBM accounts for 54% of all gliomas, and initial treatment often consists of surgery, radiation therapy, and systemic therapy18,19. Only one-third of patients will survive for 1 year, and <5% of patients will live beyond 5 years18. Given the response to currently available treatments, novel approaches are needed to treat patients with these types of cancers.
Rovalpituzumab tesirine (Rova-T) is a first-in-class antibody-drug conjugate that targets DLL3 to deliver a cytotoxic compound directly to tumor cells. Rova-T is composed of a monoclonal DLL3 antibody linked to a DNA intercalating agent (pyrrolobenzodiazepine) via a protease-cleavable linker. In SCLC and large cell neuroendocrine patient-derived xenograft models, Rova-T significantly inhibited tumor growth compared with standard platinum-based therapy by effectively targeting and eliminating DLL3-positive tumor-initiating cells2. A phase I study (NCT01901653) demonstrated Rova-T antitumor activity in patients with recurrent SCLC20. The objective response rate (ORR) was 31%, and the 1-year survival rate was 32%; the median OS was 5.8 months in patients with tumors expressing a high level of DLL3. In the phase II TRINITY study (NCT02674568) of Rova-T in patients with relapsed/refractory SCLC, the ORR was 14%, the median PFS was 3.8 months, and the median OS was 5.7 months in patients with tumors expressing a high-level DLL321.
Given the activity of Rova-T observed in studies of SCLC and the prevalence of DLL3 expression in solid tumors described above, this study examined the safety, tolerability, and antitumor activity of Rova-T in patients with DLL3-positive tumors, including NEC/NET, melanoma, MTC, GBM, and other solid tumors.
Results
Patient demographics, baseline characteristics, and disposition
Between September 2016 and February 2019, 1293 patients were pre-screened for DLL3-positive tumor status (Supplementary Table 1); ~287 patients were ultimately screened for the study, and 200 patients were subsequently enrolled and received at least one dose of Rova-T. Disease-specific cohorts included both a dose-escalation and expansion cohort. As of the data cutoff of October 14, 2019, patients had received a median of one cycle of therapy (range, 1‒5); 96 patients (48%) had received two or more cycles of Rova-T.
There were 101 patients with NEC/NET (pulmonary and extrapulmonary large cell NEC [n = 13], NEPC [n = 21], high-grade gastroenteropancreatic [GEP] NEC [n = 36], and other NEC/NET [n = 31]) and 99 patients with other solid tumors (melanoma [n = 20], MTC [n = 13], GBM [n = 23], and other [n = 43]). Supplementary Table 2 provides a breakdown of primary diagnosis for other NEC/NET. The median age was 61 (range, 28‒84) years, and 94% of patients had stage IV disease at study entry (Table 1). Seventy-seven (39%) patients had tumors expressing a high level of DLL3, which was defined as ≥50% DLL3-positive cells. Most patients (55%) had received three or more prior therapies. The median duration of follow-up was 4.6 (range, 0.1–33.7) months in all patients and 4.7 (range 0.1–27.1) months in patients treated at the recommended phase 2 dose (RP2D) of 0.3 mg/kg. The experience of one patient was described previously22.
Rova-T was administered at dose levels of 0.2 mg/kg (n = 43), 0.3 mg/kg (n = 145), and 0.4 mg/kg (n = 12). Reasons for discontinuation of Rova-T included disease progression (n = 58, 29%), adverse events (AEs) (n = 48, 24%), investigator decision (n = 21, 11%), withdrawn consent (n = 24, 12%), and other reasons (treatment completed: n = 14, 7%; clinical progression: n = 10, 5%; withdrawn for hospice or other treatment: n = 3, 2%; lost to follow-up: n = 2, 1%; noncompliance: n = 2, 1%; death: n = 1, 1%; and unknown reasons: n = 17, 9%).
Dose escalation findings: dose-limiting toxicities (DLTs) and RP2D
Overall, seven DLTs were experienced by five patients in this study. At the 0.2-mg/kg dose level, two of 43 (5%) patients experienced DLTs, including one patient with grade 3 photosensitivity reaction and one patient with grade 3 dyspnea. Two of 145 (3%) patients in the 0.3-mg/kg dose level had DLTs, including one patient with grade 2 effusion and one patient with grade 3 rhabdomyolysis, grade 3 tumor lysis syndrome, and grade 4 kidney injury. One of 12 (8%) patients treated at the 0.4-mg/kg dose level had a DLT of grade 4 thrombocytopenia. Despite only one DLT identified in the 0.4-mg/kg group, the safety data in totality indicated that 0.4 mg/kg every six weeks (q6w) is not well tolerated (Table 2). Of the 12 patients enrolled in that cohort, nine of 12 (75%) patients had grade 3/4 AEs, and six experienced drug-related serious AEs (SAEs), including one patient with grade 5 hepatic failure. Because of these safety findings, 0.3 mg/kg q6w for two cycles was chosen as the RP2D.
Clinical safety
Most patients (144 of 145; 99%) treated with 0.3 mg/kg Rova-T had at least one treatment-emergent AE (TEAE) (Table 3). The most common all-grade TEAEs were fatigue in 75 patients (52%), nausea in 53 patients (37%), and thrombocytopenia and pleural effusion in 48 patients (33%) each. Grade 3/4 AEs occurred in 78 of 145 (54%) patients. Fifty-nine (42%) patients had SAEs (excluding malignant neoplasm progression), most commonly pleural effusion (n = 7; 5%), pericardial effusion (n = 6; 4%), and dyspnea (n = 5; 3%; Supplementary Table 3). TEAEs of special interest were pleural effusion (n = 48; 33%), peripheral edema (n = 44; 30%), pericardial effusion (n = 38; 26%), photosensitivity reaction (n = 37; 26%), and pneumonitis (n = 3; 2%). For each of these, grade 3/4 events occurred in <5% of the overall population (Supplementary Table 4). Overall, 31 (21%) patients discontinued treatment due to TEAEs.
At the time of data cutoff, a total of 21 of 145 (14%) patients who were treated at the 0.3-mg/kg dose level experienced a grade 5 TEAE; 14 of 145 (10%) patients had a grade 5 event of malignant neoplasm progression, and one of 145 (<1%) patients had a grade 5 event of another malignancy. Six of 145 (4%) patients experienced a TEAE leading to death that was not related to disease progression or malignancy; grade 5 TEAEs that were not related to disease progression or malignancy included two events of pneumonitis, and one event each of multiple organ dysfunction, acute respiratory failure, hepatic encephalopathy, device-related infection, and acute kidney injury (Supplementary Table 5). Four of 145 (3%) patients had AEs leading to death that were related to Rova-T, including two who died of pneumonitis, one who died of acute respiratory failure, and one who died of hepatic encephalopathy.
Efficacy
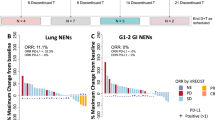
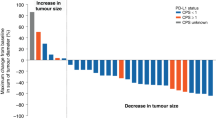
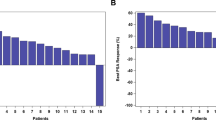
One hundred forty-five patients received at least one dose of Rova-T at 0.3 mg/kg and were included in the efficacy analyses (Table 4). The median follow-up for patients treated at 0.3 mg/kg was 4.7 months (range, 0.1–27.1). Overall, the ORR was 10%, including one complete response (CR) and 14 partial responses (PRs), and the best overall response (BOR) rate was 17% (25/145; one CR and 24 PRs). In pooled patients with NEC/NET, the ORR was 13% (9/69; all PRs) and the BOR rate was 25% (17/69; all PRs) (Supplementary Table 6). The median PFS was 4.1 months (95% CI, 2.8–4.8), and the median OS was 7.1 months (95% CI, 5.6–9.7) in pooled patients with NEC/NET. Efficacy for 43 patients treated with Rova-T at 0.2 mg/kg and 12 patients treated at 0.4 mg/kg is reported in Supplementary Tables 7 and 8. The best change in tumor lesion size in patients in each cohort is shown in Supplementary Figs. 1–8.
In pooled patients with NEC/NET expressing a high level of DLL3 (≥50% DLL3-positive tumor cells), the ORR was 17% (6/35) and 34% (12/35) had a BOR (all PRs). In those with NEC/NET expressing a low level of DLL3 (1–49% DLL3-positive tumor cells), the ORR was 9% (3/34) and the BOR rate was 15% (5/34) (all PRs; Table 5). The median PFS values for pooled patients with NEC/NET expressing high and low levels of DLL3 were 4.3 months (95% CI, 2.7–6.1) and 3.3 months (95% CI, 2.4–4.8), respectively. The median OS values for patients expressing high and low levels of DLL3 were 7.4 months (95% CI, 5.6–13.1) and 7.1 months (95% CI, 4.3–9.9), respectively.
Discussion
In this multicenter, open-label, phase I/II study, the safety and efficacy of Rova-T monotherapy were evaluated at three dose levels across advanced solid tumors with DLL3 expression. Patients treated at the 0.2-mg/kg and 0.3-mg/kg dose levels had fewer drug-related SAEs (21% and 23%, respectively) compared with those treated at 0.4 mg/kg (50%). Patients treated at the lower dose levels also had fewer drug-related TEAEs leading to death (0–3%) compared with those treated at 0.4 mg/kg (17%). Accordingly, the RP2D of Rova-T was chosen as 0.3 mg/kg q6w for two cycles with the option of treatment beyond two cycles or retreatment upon progression based on individual risk-benefit balance. The TEAEs reported in this study were similar to those observed previously with Rova-T in patients with relapsed/refractory SCLC20,21. High rates of pleural effusion, peripheral edema, pericardial effusion, and photosensitivity were observed in patients treated at 0.3 mg/kg. The rate of any-grade pericardial effusion in this study (26%) was higher than reported in previous studies of Rova-T (14–16%)20,21. Grade 3/4 TEAEs of special interest occurred at low rates. Overall, Rova-T at 0.3 mg/kg q6w had a more manageable toxicity profile than did 0.4 mg/kg for the majority of patients, and no appreciable differences in toxicity profiles were observed across the tumor types evaluated. Owing to the small sample sizes, it is not possible to conclude whether small differences in the toxicity profiles between different tumor types or between the 0.2 mg/kg and 0.3 mg/kg dose levels were statistically significant; however, the 0.4 mg/kg dose was clearly more toxic. Strategies to mitigate toxicity that were utilized in this study included premedication with steroids, educating investigators on important risks (pleural/pericardial effusions, edema, photosensitivity, and pneumonitis), reminding investigators to closely monitor for risks and manage with standard clinical practice, and sharing of best practices among investigators.
Toxicities associated with Rova-T treatment, including pleural and pericardial effusions, may be caused in part by the pyrrolobenzodiazepine component of the antibody-drug conjugate. While the mechanism is not fully understood, studies suggest that systemic release or bystander effect may be involved22,23,24. Systemic release occurs when premature cleavage of the linker results in the release of the drug into circulation, causing off-target toxicities. Bystander effect is the diffusion of the drug from the target cell to neighboring cells that do not have the target protein, either by leaking from the targeted cell or cleavage of the drug before it is internalized. In either case, DLL3-negative cells may be inappropriately exposed to pyrrolobenzodiazepine, and further refinement of the linker or the drug-antibody ratio may mitigate these effects.
This study enrolled a heavily pretreated population of patients with DLL3-expressing tumors, and 55% of patients received at least three prior lines of therapies. These findings demonstrate that Rova-T as a single agent had antitumor activity in a subset of heavily pretreated patients with tumor types that express DLL3, including NEC/NET, melanoma, and MTC. These tumor types tend to be treatment refractory following multiple prior lines of therapy, and this may have contributed to the low response rate. In addition, although DLL3 expression by IHC was required for study entry, intratumoral variability in DLL3 expression may have limited the overall antitumor activity of Rova-T. The median PFS and OS in the patients in this study with the refractory disease were similar to ranges shown in previous studies25,26,27,28.
Despite the limited sample size, a trend toward a higher response rate was observed among patients with NEC/NET with high DLL3 expression, supporting DLL3 as a promising therapeutic target for the treatment of NEC/NET across primary disease sites, including treatment-emergent NEPC. For pooled patients with NEC/NET, the duration of response (DOR) of 3.1 months appears comparable to the efficacy with common treatment for these tumor types in the relapsed or refractory setting29. These findings are of particular interest for patients with recurrent NEC, for which there is no standard-of-care treatment. Of note, the category of grade 3 NET was not established for pancreatic and gastrointestinal NET for most of the study enrollment, therefore it is not known whether any patients with grade 3 GEP NET were part of the study. The majority of patients who were enrolled in the other NEC/NET cohort were classified as having NEC, so it was not possible to determine whether there was a difference in DLL3 expression in NET versus NEC.
Overall, refinement of both the drug-antibody ratio and the linker in Rova-T could improve drug delivery, reduce toxicity, and increase treatment duration, which could lead to improved efficacy. Further study is needed to define the risk-benefit balance of Rova-T in patients with NEC/NET. During the conduct of the current trial, further development of Rova-T was discontinued based on results from two phase III studies that indicated a lack of favorable risk-benefit balance of Rova-T in patients with SCLC (https://news.abbvie.com/news/press-releases/abbvie-discontinues-rovalpituzumab-tesirine-rova-t-research-and-development-program.htm; https://news.abbvie.com/news/phase-3-trial-rova-t-as-second-line-therapy-for-advanced-small-cell-lung-cancer-tahoe-study-halted.htm). However, DLL3 remains a relevant anticancer target. Additional strategies targeting DLL3 in these difficult-to-treat tumor types warrant investigation.
Methods
Patients
Adult patients with unresectable, refractory, advanced solid tumors other than SCLC who were positive for DLL3 and had measurable disease were included in the study. DLL3 positivity was defined as immunohistochemical staining in ≥1% of tumor cells. Potential patients were pre-screened for DLL3 positivity to determine initial eligibility, and those with DLL3-positive tumors underwent full screening for study eligibility upon disease progression. Measurable disease was defined based on Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1)30. Patients had to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 1, a life expectancy of ≥12 weeks, and satisfactory laboratory parameters. Patients could not have a clinically significant medical condition, including uncontrolled hypertension and/or diabetes, pulmonary disease, neurological disorder, recent or ongoing serious infection, or a cerebral vascular event within six months of starting the study. Prior exposure to pyrrolobenzodiazepine-containing drugs, including Rova-T, was not allowed. All patients provided written informed consent.
Study design and objectives
This multicenter, open-label, phase I/II study (NCT02709889) enrolled patients in the United States. The study was conducted according to the Declaration of Helsinki and all applicable laws, rules, and regulations within the relevant jurisdictions of the investigators; the study was approved by the institutional review boards at each participating institution. Patients were enrolled in disease-specific cohorts, including melanoma, MTC, GBM, large-cell NEC, NEPC, GEP NEC, other NEC/NET, and other solid tumors. Dosing was predetermined to start at 0.2 mg/kg q6w, which is one dose level below the RP2D for SCLC, with dose escalation continuing through 0.4 mg/kg, the maximum tolerated dose for SCLC20. In Part A, the RP2D was determined in disease-specific cohorts. Part B tested the RP2D determined in Part A in disease-specific expansion cohorts. The primary endpoint was safety. Secondary endpoints included BOR, ORR, DOR, PFS, and OS. The relationship between DLL3 expression and clinical outcome was tested as an exploratory endpoint.
Treatment and assessments
DLL3 expression was determined at baseline with fresh or archived tumor tissue using an SC16.65 mouse antibody IHC investigational use only (IUO) assay developed by Ventana Medical Systems as previously described, at a concentration of 0.78 µg/ml (AbbVie Stemcentrix, Lot No. 170420)20,21. Rova-T was administered intravenously on day 1 of each 6-week cycle at 0.2, 0.3, or 0.4 mg/kg with a 3 + 3 design for dose escalation in Part A. Patients received treatment until disease progression or unacceptable toxicity in Part A. Patients in Part B received two doses and could receive further doses at the discretion of the investigator. Dosing interval and duration were selected based on previous clinical studies of Rova-T in SCLC20,21. Dexamethasone (8 mg) was administered orally twice daily on the day before treatment with Rova-T and on days 1 and 2 of treatment in each cycle. Rova-T dose reductions were allowed. DLTs were evaluated in the DLT evaluation period, which occurred during the first three weeks of the first cycle of treatment. DLTs were defined as grade 4 thrombocytopenia (or grade 3 with bleeding) lasting >7 days or requiring platelet transfusion, grade 4 neutropenia lasting >7 days and/or requiring growth factor support, any febrile neutropenia, grade 4 anemia unrelated to underlying disease, clinically significant grade 3/4 nonhematologic laboratory abnormalities lasting >7 days, and grade 3/4 nonlaboratory AEs with the exception of fatigue, asthenia, nausea, or other constitutional symptoms. Grade ≥3 AEs clearly unrelated to study drug and grade ≥3 AEs of isolated alkaline phosphatase, amylase, or lipase laboratory abnormalities were not considered DLTs.
Disease assessments involved computed tomography (CT) scans of the chest, abdomen, pelvis, and neck (if indicated) and were conducted q6w during active study treatment for 24 weeks and every 12 weeks thereafter until disease progression. MRI scans of the brain were conducted if central nervous system progression was previously documented, and CT scans with intravenous contrast could be substituted at the discretion of the investigator. Patients with prostate cancer underwent whole-body technetium-99m bone scintigraphy. Tumor response was assessed by investigators according to RECIST v1.1, Response Assessment in Neuro-Oncology criteria for GBM, and Prostate Cancer Clinical Trials Working Group 3 (PCWG3) for prostate cancer30. AEs were summarized using preferred terms from the Medical Dictionary for Regulatory Activities and graded using the National Cancer Institute’s Common Terminology Criteria for Adverse Events version 4.03.
Statistical analysis
The planned enrollment was ~144 patients in dose escalation and ~174 patients in dose expansion to detect an ORR of 15%, which would indicate efficacy worthy of further investigation. Efficacy was assessed by disease and cohort and included all patients who received at least one dose of Rova-T. BOR (defined as the best response of CR or PR, with confirmation not required), ORR (confirmed response), and DOR were summarized for all patients with a CR or PR according to RECIST v1.1 or PCWG3. DOR, PFS, and OS were evaluated using the Kaplan–Meier method. For DOR and PFS, patients were censored at the time at which they received another cancer therapy, missed two tumor assessments in a row, or had their last evaluable response assessment if not PD or death. Safety assessments were performed in all patients who received at least one dose of Rova-T.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual, and trial-level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time, and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
References
Chapman, G., Sparrow, D. B., Kremmer, E. & Dunwoodie, S. L. Notch inhibition by the ligand DELTA-LIKE 3 defines the mechanism of abnormal vertebral segmentation in spondylocostal dysostosis. Hum. Mol. Genet. 20, 905–916 (2011).
Saunders, L. R. et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci. Transl. Med. 7, 302ra136 (2015).
von Arx, C. et al. Updates on the role of molecular alterations and NOTCH signalling in the development of neuroendocrine neoplasms. J. Clin. Med. 8, https://doi.org/10.3390/jcm8091277 (2019).
Lim, J. S. et al. Intratumoural heterogeneity generated by Notch signalling promotes small-cell lung cancer. Nature 545, 360–364 (2017).
Dunwoodie, S. L., Henrique, D., Harrison, S. M. & Beddington, R. S. Mouse Dll3: a novel divergent Delta gene which may complement the function of other Delta homologues during early pattern formation in the mouse embryo. Development 124, 3065–3076 (1997).
Hermans, B. C. M. et al. DLL3 expression in large cell neuroendocrine carcinoma (LCNEC) and association with molecular subtypes and neuroendocrine profile. Lung Cancer 138, 102–108 (2019).
Puca, L. et al. Delta-like protein 3 expression and therapeutic targeting in neuroendocrine prostate cancer. Sci. Transl. Med. 11, https://doi.org/10.1126/scitranslmed.aav0891 (2019).
Zhang, P. et al. Etoposide and cisplatin versus irinotecan and cisplatin as the first-line therapy for patients with advanced, poorly differentiated gastroenteropancreatic neuroendocrine carcinoma: a randomized phase 2 study. Cancer 126, 2086–2092 (2020).
Yamaguchi, T. et al. Multicenter retrospective analysis of systemic chemotherapy for advanced neuroendocrine carcinoma of the digestive system. Cancer Sci. 105, 1176–1181 (2014).
Mitry, E. et al. Treatment of poorly differentiated neuroendocrine tumours with etoposide and cisplatin. Br. J. Cancer 81, 1351–1355 (1999).
Hainsworth, J. D., Spigel, D. R., Litchy, S. & Greco, F. A. Phase II trial of paclitaxel, carboplatin, and etoposide in advanced poorly differentiated neuroendocrine carcinoma: a Minnie Pearl Cancer Research Network study. J. Clin. Oncol. 24, 3548–3554 (2006).
Sorbye, H. et al. Predictive and prognostic factors for treatment and survival in 305 patients with advanced gastrointestinal neuroendocrine carcinoma (WHO G3): the NORDIC NEC study. Ann. Oncol. 24, 152–160 (2013).
Welin, S. et al. Clinical effect of temozolomide-based chemotherapy in poorly differentiated endocrine carcinoma after progression on first-line chemotherapy. Cancer 117, 4617–4622 (2011).
Spino, M. et al. Cell surface notch ligand DLL3 is a therapeutic target in isocitrate dehydrogenase-mutant glioma. Clin. Cancer Res. 25, 1261–1271 (2019).
Dummer, R. et al. Cutaneous melanoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 26, v126–v132 (2015).
Robert, C. et al. Pembrolizumab versus ipilimumab in advanced melanoma. N. Engl. J. Med. 372, 2521–2532 (2015).
Randle, R. W. et al. Trends in the presentation, treatment, and survival of patients with medullary thyroid cancer over the past 30 years. Surgery 161, 137–146 (2017).
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2006-2010. Neuro. Oncol. 15, ii1–ii56 (2013).
Stupp, R. et al. High-grade glioma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 25, iii93–iii101 (2014).
Rudin, C. M. et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: a first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 18, 42–51 (2017).
Morgensztern, D. et al. Efficacy and safety of rovalpituzumab tesirine in third-line and beyond patients with DLL3-expressing, relapsed/refractory small-cell lung cancer: results from the phase II TRINITY study. Clin. Cancer Res., https://doi.org/10.1158/1078-0432.CCR-19-1133 (2019).
Miller, M. L. et al. A DNA-interacting payload designed to eliminate cross-linking improves the therapeutic index of antibody-drug conjugates (ADCs). Mol. Cancer Ther. 17, 650–660 (2018).
Joubert, N., Beck, A., Dumontet, C. & Denevault-Sabourin, C. Antibody-drug conjugates: the last decade. Pharmaceuticals 13, https://doi.org/10.3390/ph13090245 (2020).
Khera, E. et al. Quantifying ADC bystander payload penetration with cellular resolution using pharmacodynamic mapping. Neoplasia 23, 210–221 (2021).
Kam Kamiya-Matsuoka, C. & Gilbert, M. R. Treating recurrent glioblastoma: an update. CNS Oncol. 4, 91–104 (2015).
Yamazaki, N. et al. Long-term follow up of nivolumab in previously untreated Japanese patients with advanced or recurrent malignant melanoma. Cancer Sci. 110, 1995–2003 (2019).
Wells, S. A. Jr. et al. Revised American thyroid association guidelines for the management of medullary thyroid carcinoma. Thyroid 25, 567–610 (2015).
Okuyama, H. et al. A phase II trial of everolimus in patients with advanced pancreatic neuroendocrine carcinoma refractory or intolerant to platinum-containing chemotherapy (NECTOR trial). Neuroendocrinology, https://doi.org/10.1159/000505550 (2020).
Sugiyama, K. et al. Salvage chemotherapy by FOLFIRI regimen for poorly differentiated gastrointestinal neuroendocrine carcinoma. J Gastrointest. Canc., https://doi.org/10.1007/s12029-020-00516-7 (2020).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Eckardt, J. R. et al. Phase III study of oral compared with intravenous topotecan as second-line therapy in small-cell lung cancer. J. Clin. Oncol. 25, 2086–2092 (2007).
Acknowledgements
AbbVie and the authors thank the patients and their families, study coordinators, and support staff, as well as the former AbbVie employees for contributions to the study. Medical writing support was provided by Rohina Rubicz, Ph.D., of Bio Connections LLC, funded by AbbVie. AbbVie sponsored the study and participated in the design, study conduct, analysis, and interpretation of data, as well as the writing, review, and approval of the publication. No honoraria or payments were made for authorship.
Author information
Authors and Affiliations
Contributions
Conception and design: H.B., R.A. Provision of study materials or patients: A.F.F., H.B., A.E.H., M.H.T., S.T.T., J.N., C.H.C., R.A. Collection and assembly of data: A.F.F., A.E.H., M.H.T., S.T.T., J.N., C.H.C., C.L., Y.L., R.A. Data analysis and interpretation: A.E.H., M.H.T., S.T.T., J.N., Y.L., R.A. Manuscript writing: all authors. Final approval of manuscript: all authors.
Corresponding author
Ethics declarations
Competing interests
A.S.M. reports honoraria to institution from AbbVie, AstraZeneca, Bristol Myers Squibb, and Genentech; research funding from the National Institutes of Health, Novartis, and Verily; nonremunerated director of Mesothelioma Applied Research Foundation. D.S.H. reports a leadership role or other ownership with OncoResponse, Molecular Match, and Presagia Inc.; a consultancy/advisory role for Alpha Insights, Amgen, Axiom, Adaptimmune, Baxter, Bayer, eCancer, Genentech, GLG, Group H, Guidepoint, Infinity, Medscape, Numab, Oncology Education Project Association, Pfizer, Prime Oncology, Takeda, Trieza Therapeutics, and WebMD; travel support from Bayer, Loxo, miRNA, Genmab, AACR, ASCO, and SITC; research funding from AbbVie, Adaptimmune, Aldi-Norte, Amgen, AstraZeneca, Bayer, Bristol Myers Squibb, Daiichi Sankyo, Eisai, Fate Therapeutics, Genentech, Genmab, GlaxoSmithKline, Ignyta, Infinity, Kite, Kyowa, Eli Lilly, Loxo Oncology, Merck, MedImmune, Mirati Therapeutics, miRNA, Molecular Templates, MOLOGEN AG, NCI-CTEP, Novartis, Pfizer, Seattle Genetics, Takeda, and Turning Point Therapeutics. C.L.H. reports a consultancy/advisory role with AbbVie, Ascentage Pharma, Bristol Myers Squibb, and Genentech; research funding to institution from AbbVie, Amgen, AstraZeneca, and Bristol Myers Squibb. A.F.F. reports a consultancy/advisory role with Bayer, Loxo Oncology, Genentech, Roche, Bristol Myers Squibb, AstraZeneca, AbbVie, PharmaMar, Boehringer Ingelheim, Merck, H3 Biomedicine, and Pfizer; research funding from Bayer, Loxo Oncology, Genentech, Roche, Bristol Myers Squibb, AstraZeneca, AbbVie, PharmaMar, Merck, Ignyta, Amgen, and Novartis; honoraria from DAVA Oncology, Clinical Care Options, Medical Learning Institute, Medscape, PeerView, and Research to Practice. H.B. reports a consultancy/advisory role with Janssen, Sanofi Genzyme, Pfizer, Astellas, AstraZeneca, and Merck; research funding from AbbVie/Stemcentrx; research funding to institution from Janssen Oncology, Eli Lilly, and Millennium Pharmaceuticals. S.N.W. reports research funding to institution from Spectrum Pharmaceuticals, Eli Lilly, Pfizer, Roche/Genentech, Daiichi Sankyo, NewLink Genetics, EMD Serono, Puma Biotechnology, Novartis, Xcovery, Synermore Biologics, Celgene, Vertex, Bristol Myers Squibb, Stemcentrx, Hengrui Therapeutics, Checkpoint Therapeutics, Ignyta, AstraZeneca, ARIAD, Roche, and Merck. A.E.H. reports a consultancy/advisory role with Novartis, Ipsen, Perthera, Celgene, and AbbVie; research funding from AbbVie; travel accommodations from Halozyme. L.B.A. reports a consultancy/advisory role for Lexicon; research funding to institution from AbbVie, Oncotelic, and Entrinsic Health Solutions. M.H.T. reports a consultancy/advisory role with and honoraria from Bristol Myers Squibb, Eisai, Novartis, Bayer, Sanofi/Genzyme, Array Biopharma, Loxo Oncology, Blueprint Medicines, and Arqule; speakers’ bureau for and honoraria from Bristol Myers Squibb and Eisai. A.H.B. reports honoraria from Astellas Pharma and Bayer; travel support from Clovis Oncology. S.T.T. reports a consultancy/advisory role for Medivation, Astellas Pharma, Dendreon, Janssen, Bayer, Genentech, Endocyte, Immunomedics, Karyopharm Therapeutics, AbbVie, Tolmar, QED Therapeutics, Amgen, Sanofi, Pfizer, Clovis Oncology, Novartis, and Genomic Health, POINT Biopharma, Blue Earth Diagnostics, Aikido Pharma, Telix Pharma; travel support from Sanofi, Immunomedics, and Amgen; research funding to institution from Eli Lilly, Sanofi, Janssen, Astellas Pharma, Progenics, Millennium, Amgen, Bristol Myers Squibb, Dendreon, Rexahn Pharmaceuticals, Bayer, Genentech, NewLink Genetics, Inovio Pharmaceuticals, AstraZeneca, Immunomedics, Novartis, AVEO, Boehringer Ingelheim, Merck, Stemcentrx, Karyopharm Therapeutics, AbbVie, Medivation, Endocyte, Exelixis, Clovis Oncology, SeaGen, and Gilead. K.L. reports a consultancy/advisory role for Array BioPharma, Merck, Roche, and Regeneron; honoraria from Array BioPharma; travel support from Merck, Roche/Genentech, and Regeneron; research funding to the institution from Roche/Genentech, Merck, Array BioPharma, Incyte, Nektar, Iovance Biotherapeutics, Bristol Myers Squibb, Kartos Therapeutics, OncoSec, Regeneron, Alkermes, and Neon Therapeutics; uncompensated relationship with Roche/Genentech and Regeneron. J.N. reports a consultancy/advisory role for Roche and Boehringer Ingelheim; honoraria from Onclive. C.H.C. reports a consultancy/advisory role for Bristol Myers Squibb, Mirati, CUE Biopharma, Sanofi, and Ignyta; travel support from Bristol Myers Squibb; research funding from AstraZeneca, Bristol Myers Squibb, Eli Lilly, Merck, Regeneron, Ignyta, Brooklyn Therapeutics, Pfizer, and lovance. J.M.C. reports research funding from Merck, AstraZeneca, Esperas Pharma, and Tesaro; a consultancy/advisory role with Bristol Myers Squibb; travel funding from Bristol Myers Squibb. M.R. reports former employment by AbbVie, current employment with Calithera Biosciences, and may hold AbbVie stock. C.L., R.V., and Y.L. report employment by AbbVie and may own stock or other options. R.A. reports a consultancy/advisory role for AstraZeneca and Janssen; honoraria from Clovis Oncology; travel support from Xynomic Pharmaceuticals; institution received research funding from Zenith Epigenetics, Novartis, Xynomic Pharmaceuticals, Cancer Targeted Technology, Janssen, Merck, and AbbVie.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mansfield, A.S., Hong, D.S., Hann, C.L. et al. A phase I/II study of rovalpituzumab tesirine in delta-like 3—expressing advanced solid tumors. npj Precis. Onc. 5, 74 (2021). https://doi.org/10.1038/s41698-021-00214-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-021-00214-y
This article is cited by
-
A systematic review of immunotherapy in high-grade glioma: learning from the past to shape future perspectives
Neurological Sciences (2024)
-
Emerging therapies targeting the delta-like ligand 3 (DLL3) in small cell lung cancer
Journal of Hematology & Oncology (2023)
-
Comprehensive analysis of necroptotic patterns and associated immune landscapes in individualized treatment of skin cutaneous melanoma
Scientific Reports (2023)
-
Repurposing ketotifen as a therapeutic strategy for neuroendocrine prostate cancer by targeting the IL-6/STAT3 pathway
Cellular Oncology (2023)
-
Potent molecular-targeted therapies for gastro-entero-pancreatic neuroendocrine carcinoma
Cancer and Metastasis Reviews (2023)
