
Abstract
The study explored the clinical significance of fetal loss of heterozygosity (LOH) identified by single-nucleotide polymorphism array (SNP array). We retrospectively reviewed data from pregnant women who underwent invasive diagnostic procedures at prenatal diagnosis centers in southeastern China from December 2016 to December 2021. SNP array was performed by the Affymetrix CytoScan 750 K array platform. Fetuses with LOH were further identified by parental verification, MS-MLPA, and/or trio whole-exome sequencing (trio-WES). The genetic results, fetal clinical manifestations, and perinatal outcome were analyzed. Of 11,062 fetuses, 106 (0.96%) had LOH exhibiting a neutral copy number, 88 (83.0%) had LOH in a single chromosome, whereas 18 (17.0%) had multiple LOHs on different chromosomes. Sixty-six fetuses had ultrasound anomalies (UAs), most frequently fetal growth restriction (18/66 (27.3%)). Parental SNP array verification was performed in 21 cases and trio-WES in 21 cases. Twelve cases had clinically relevant uniparental disomy, five had pathogenic variants, four had likely pathogenic variants, six had variants of unknown significance, and eight had identity by descent. The rate of adverse pregnancy outcomes in fetuses with LOH and UAs (24/66 (36.4%)) was higher than in those without UAs (6/40 (15.0%)) (p < 0.05). LOH is not uncommon. Molecular genetic testing techniques, including parental SNP array verification, trio-WES, methylation-specific multiplex ligation-dependent probe amplification, regular and systematic ultrasonic monitoring, and placental study, can accurately assess the prognosis and guide the management of the affected pregnancy.
Similar content being viewed by others
Introduction
In prenatal diagnostics, chromosomal microarray analysis (CMA) improved the diagnostic rate for chromosomal abnormalities by 4.1–10% compared with traditional karyotyping in fetuses with ultrasound anomalies (UA). Single-nucleotide polymorphism array technology can not only identify copy number variation (CNV) but also detect chromosome aneuploidy and haploidy, triploidy, loss of heterozygosity (LOH), uniparental disomy (UPD), and low-level mosaicism1,2.
LOH, referred to as the region of homozygosity in a chromosome, and the clinical implications, are associated concerns for identity by descent (IBD), consanguinity, UPD, and recessive single-gene mutations. LOH confirmed to have been inherited from only one parent is called UPD, which can lead to imprinting disorders involving imprinted chromosomes 6, 7, 11, 14, 15, and 20. Moreover, UPDs are classified as either isodisomy, heterodisomy, or mixed UPD, according to the parental origin. Common mechanisms resulting in UPD include trisomy rescue, monosomy rescue, and somatic mitotic recombination, resulting in mosaic segmental UPD3,4.
Assessing the prognosis of fetuses with LOH in prenatal diagnosis is challenging. Thus, understanding its clinical significance is necessary due to the phenotypic complexity of LOH and its uncertain pathogenicity. To assess the clinical significance and better understand the correlation between LOH and its phenotype, we investigated the clinical manifestations; performed further molecular genetic analysis using parental SNP array verification, trio whole-exome sequencing (WES), and methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA); and tracked the perinatal outcomes of fetuses with LOH.
Patients and methods
Subjects
We retrospectively reviewed pregnant women undergoing invasive diagnostic procedure for a variety of indications at all prenatal diagnosis centers in Fujian Province in southeastern China, from December 2016 to December 2021. Most of the cases came from Fujian Maternity and Child Health Hospital. Obtaining informed consent from all the pregnant couples, fetal samples were collected via invasive diagnostic procedure according different weeks of gestation. The study was approved by the Ethics Committee of the Fujian Maternity and Child Health Hospital (No.2016KYLLD01051).
Conventional karyotyping analysis
Karyotyping was performed following the standard cytogenetic protocol5, and karyotypes were scanned on Leica GSL120. At least 20 metaphases were counted, and five metaphases were analyzed. Karyotype analysis and description were based on ISCN 20206.
Isolation of genomic DNA
Fetal sample, 15 mg of chorionic villi, 30–40 mL of amniotic fluid, or 2–5 mL umbilical cord blood was obtained, and genomic DNA from the fetus and its parents were extracted using the QIAamp® DNA Blood Mini Kit (Qiagen Inc., Hilden, Germany) following the manufacturer’s instructions, and maternal cell contamination was ruled out using multiplex quantitative-fluorescent polymerase chain reaction Darui kit (Darui, Guangzhou, China), which was tested on 20 markers including: four short tandem repeats (STRs) from chromosome 13 (D13S634, D13S305, D13S628, D13S742), four from chromosome 18 (D18S391, D18S1002, D18S535, D18S386), six from chromosome 21 (D21S1411, D21S1445, D21S1414, D21S1412, D21S1433, 21q11.2), and six STRs from chromosome X and Y (AMXY, DXS1187, DXS8377, SRY, DXS6809, DXS981).
Single nucleotide polymorphism array and data analysis
Chromosomal aberrations, CNVs and LOH were detected using a SNP array on a CytoScan 750 K (Affymetrix Inc., Santa Clara, CA) platform, all the experimental processes of SNP array were performed as previously described7. Parental SNP array verification was performed to confirm the origin of fetal LOH.
The raw data were analyzed using the Affymetrix Chromosome Analysis Suite software (version 3.1.0.15). The coordinate of the chromosome was described based on the genome version hg19. CNVs were classified according to the American College of Medical Genetics (ACMG) guidelines8. The reporting threshold was set at CNV ≥ 500 Kb, and LOHs cutoffs of > 5 Mb for telomeric LOH and/or > 15 Mb for interstitial LOH on imprinted chromosomes (chromosome 6, 7, 11, 14, 15, and 20); > 10 Mb for telomeric LOH and/or > 20 Mb for interstitial LOH on nonimprinted chromosomes; > 15 Mb for interstitial LOH on imprinted chromosomes or 20 Mb for an interstitial LOH on nonimprinted chromosomes; percentage of LOH was larger than or equal to 1.625%9. The sex chromosomes are excluded, because males have one X and one Y chromosome and therefore cannot have LOH at any site apart from the pseudoautosomal regions10. The significance of LOH results was interpreted through PubMed (http://www.ncbi.nlm.nih.gov/pubmed), Online Mendelian Inheritance in Man (OMIM; http://www.omim.org/), DECIPHER (https://decipher.sanger.ac.uk/), UCSC (http://genome.ucsc.edu/), ClinGen Dosage Sensitivity Map (https://www.ncbi.nlm.nih.gov/projects/dbvar/clingen/index.shtml), uniparental disomy (http://cs-tl.de/DB/CA/UPD/0-Start.html), Geneimprint (http://www.geneimprint.com/), the Catalogue of Imprinted Genes (www.otago.ac.nz/IGC), the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php), the Locus-Specific Mutation Database (http://www.hgvs.org/dblist/glsdb.htm) and the 1000 Genomes Project Dataset (https://www.ncbi.nlm .nih.gov/variation/tools/1000genomes/).
WES and bioinformatics analysis
To identify homozygous mutations of recessive diseases in addition to UPD, trio-WES (for fetuses and parents) was carried out with the informed consent of pregnant couples. After the sample genomic DNA was extracted, exons were captured using Agilent Sure Select Technology (Agilent, Santa Clara, CA, USA), fragmented randomly, purified, and enriched to construct DNA libraries. Paired-end sequencing was performed on Illumina HiSeq 2500 (Illumina, USA) instruments according to the manufacturer’s instructions (Illumina, San Diego, CA, USA).
For sequence alignment, variant calling, and annotation, the sequences were mapped to their locations with the human genome reference sequence (hg19 build) using Burrows-Wheeler software (version 0.59)11. All SNVs and InDels were annotated with public population frequency databases, including NCBI dbSNP, 1000 Genomes Project, the Exome Aggregation Consortium, as well as OMIM, Swiss-var, Human Gene Mutation Database, ClinVar, and other disease databases, and only variants that were clinically or potentially relevant to the patients’ phenotype were reported. Mutations were annotated, protein function effects and shear harmfulness were predicted, and the pathogenicity of the variants was assessed according to ACMG12.
MS-MLPA
Methylation analysis of 7q21.13q36.3 loci was performed using MS-MLPA (SALSA MS-MLPA probe mix ME030-C3 BWS/RSS; MRC Holland, Amsterdam, The Netherlands), and the relative copy numbers of the three methylation probes in the MEST gene (maternal methylation gene region, with paternal methylation preferentially expressed) on chromosome 7q32.2 was determined. All procedures were performed following the manufacturers’ protocols as previously described13.
Statistical analysis
In fetuses with LOH, we collected data relating to basic information, imaging findings, serological Down's screening results, non-invasive prenatal testing results, invasive diagnostic testing results, further genetic analysis, perinatal outcomes, and follow-up information. Perinatal outcomes in our hospital were obtained from delivery records. Data relating to cases from other centers were followed up via telephone.
Ethics approval and consent to participate
SPSS software version 22.0 was used for statistical analysis. Measurement data were expressed as mean ± standard deviation, statistical comparisons were performed using χ2 test, and p < 0.05 was considered statistically significant.
Ethics approval and consent to participate
The study complied with the principles set forth in the Declaration of Helsinki. It was approved by the Institutional Review Board of Fujian Maternal and Child Health Hospital. Written informed consent was obtained from each patient.
Results
Patient characteristics
In total, 11,062 fetuses undergoing invasive diagnostic testing over a period of five years were analyzed using SNP array, and 106 fetuses had LOH. The detection rates of fetal LOH for different invasive diagnostic indications are shown in Table 1. The mean weeks of gestation at invasive prenatal diagnosis and maternal age for pregnancies with fetal LOH were 21 ± 1 (range, 11+6 to 31) and 31 ± 3 (range, 19–42) years, respectively. The detailed parental SNP array verification results, trio-WES results, MS-MLPA results, ultrasound findings, and perinatal outcomes of 42 fetuses with LOH are summarized in Table 2, and the data regarding the remaining 64 prenatally diagnosed cases with LOH that declined further genetic testing are listed in Table 3.
Fetal LOH detected by SNP array
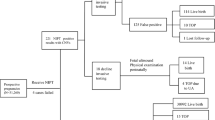
The overall flow of fetal LOH analysis is illustrated in Fig. 1. The detection rate of fetuses with LOH was 0.96% (106/11,062). In 88 (83.0%) fetuses, LOH occurred on a single chromosome, whereas in 18 (17.0%) fetuses, multiple LOHs were detected on different chromosomes. Of the 18 cases with multiple LOHs, two cases (Cases 1 and 64) were confirmed from consanguineous couples, and the remaining 16 cases denied consanguineous marriage.

The flow chart of fetal LOH analysis in our cohort. IBD, identity by descent; LOH, loss of heterozygosity; SNP array, single nucleotide polymorphism array; UPD, uniparental disomy; VOUS, variant of unknown significance; WES, whole exome sequencing.
In single-chromosome LOH, the most frequently involved chromosomes were chromosomes 6 (11.4% (10/88)), 3 (10.2% (9/88)), and 5 (8.0% (7/88)), followed by chromosomes 15 (6.8% (6/88)), 2 (6.8% (6/88)), 8 (6.8% (6/88)), and 13 (6.8% (6/88)) (Tables 2 and 3). LOH involved almost the entire chromosome in seven cases, and the involved chromosomes were chromosomes 2, 6, 13, 14, 18, and 22. In other cases, LOH occurred in only part of the chromosome, ranging from 5.18 to 96.8 Mb. In total, four cases of mosaic LOH were identified, including chromosomes 13 and 14, 5q11.1q35.3, and 7q21.13q36.3, respectively. The mosaicism rate ranged from 30 to 80%.
Both cases (Cases 22 and 28) with LOH on the entire chromosome 6 presented with fetal growth restriction (FGR), and were further diagnosed with paternal and maternal UPD6, respectively. Both patients elected termination of pregnancy (TOP) (Table 2). Isolated segmental LOH was identified on chromosome 6 in 10 cases, of which nine presented with UAs, including FGR, thickened nuchal translucency (NT), enhanced bowel echo, intracardiac echogenic focus, increased umbilical artery resistance index, oligohydramnios, cervical lymphatic hygroma, mild regurgitation of tricuspid valve, fetal bilateral renal enlargement, increased renal echogenicity, reverse a-wave of ductus venosus, and enhanced intestinal echo, resulting in TOP (n = 6) and preterm birth (n = 1). The other three cases had a favorable outcome.
Isolated LOH on chromosome 3 was identified in nine cases, of which only two presented with UAs, including thickened NT, resulting in spontaneous abortion (n = 1). The other seven cases showed no anomalies on prenatal ultrasound and had no obvious abnormal phenotypes after birth.
Isolated fetal LOH on chromosome 5 was identified in seven cases, of which three had abnormal ultrasound findings, including FGR; small fetal head circumference (HC) for gestational age; lethal bone dysplasia (osteogenesis imperfecta type II); micrognathia; small biparietal diameter (BPD), HC, and femur length (FL) for gestational age; bilateral femoral curvature; less than the normal predictive value -2SD; hydrops fetalis; fetal giant bladder; and fetal lung cystic adenoma, resulting in TOP (n = 3) or preterm birth (n = 1), and missed abortion (n = 2); the other case (Case 34) showed minor abnormal phenotypes on prenatal ultrasound, and presented micrognathia after birth.
UAs in fetal LOH detected by SNP array
In total, 66 fetuses presented UAs, including 22 (33.3%) with structural abnormalities, 24 (36.4%) with ultrasonic soft marker, 18 (27.3%) with FGR and nine (13.6%) with other presentations. The most common soft marker anomaly was thickened NT. The most frequent ultrasonic structural anomalies were cardiovascular (9.1%), skeletal (6.1%), and genitourinary malformations (6.1%). Fetal LOH mostly involved chromosomes 5, 6, and 1 in the group with UAs, and chromosomes 3, 10, and 18 in the group without UAs (Tables 2 and 3).
FGR was detected prenatally in 18 fetuses, of which 11 had FGR as an isolated ultrasound finding. The most frequently involved chromosome was chromosome 6 (n = 6) in cases with FGR, followed by chromosomes 7 (n = 3) and 17 (n = 3). The outcomes of these 18 fetuses included TOP (n = 11), preterm birth (n = 3), and term birth (n = 4). The incidence of TOP was significantly higher in fetuses with FGR than in those without FGR (61.1% (11/18) versus 21.6% (19/88), p < 0.01) (Tables 2 and 3).
UPD results
To verify the parental source of fetal LOH, 21 cases of fetal LOH were confirmed by parental SNP array analysis, of which two (9.5%) had paternal UPD (Cases 4 and 22) and 12 (57.1%) had maternal UPD (Cases 4 (UPD 14 pat), 6 (UPD7 mat), 14 (mosaic UPD7 mat), 19 (UPD15 mat), 21 (UPD 16 mat), 22 (UPD6 pat), 24 (UPD15 mat), 25 (UPD11 mat), 26 (UPD7p22.3p12.2 mat), and 30 (UPD11 mat)); four cases were confirmed to have IBD (Cases 36–42). Among the 14 cases with UPD, Cases 19 and 24 were diagnosed with Prader–Willi syndrome (PWS), Cases 6, 14, 25, 26, and 30 were determined to have Silver-Russell syndrome (SRS), and Cases 4 and 22 were diagnosed with Kagami-Ogata syndrome (KOS) and transient neonatal diabetes mellitus, respectively. No confirmed imprinted genes were detected in Cases 20, 23, 27, and 28, and UPD16 mat was found in Case 21, thus classifying them as variants of uncertain significance (VOUS). Among the 14 UPDs, notably, amniocentesis was performed in Case 14, due to a fetal right aortic arch with aberrant left subclavian artery detected on ultrasound, and LOH with a size of 70.4 Mb was observed in 7q21.13q36.3 by SNP array (Fig. 2A). First, parental SNP array verification indicated that it was not possible to determine whether the source of the LOH on chromosome 7 was paternal or maternal (Fig. 2B); then, MS-MLPA for the methylation analysis of 7q21.13q3615 loci revealed that the relative copy numbers of the three methylation probes (184, 190, and 256 bp) in the MEST gene (maternal methylation gene region, paternal methylation was preferentially expressed) were 0.66, 0.66, and 0.64, respectively, suggesting a possible low proportion mosaic maternal UPD7 associated with Silver-Russell syndrome (Fig. 2C). However, the experimental result was close to the threshold range; thus, it could not be determined and interpreted accurately. The fetus was delivered at term and had feeding difficulties after birth, whereas three months after delivery, the child's height and development were normal, except for light weight (Table 2).


(A) 70.4 Mb loss of heterozygosity (LOH) was detected by SNP array (A), and further confirmed as maternal uniparental disomy 7 (UPD7) (B and C) for case 14. Detection of a 70.4 Mb segmental LOH on 7q21.13q36.3 using a SNP array. Purple bars represent stretches of LOH occurring on 7q21.13q36.3. The allele difference panel indicates the genotype for each SNP. Confirmation of low proportion mosaic maternal UPD7 using the methylation-specific multiplex ligation dependent probe amplification (MS-MLPA). MS-MLPA revealed that the relative copy numbers of the three methylation probes in MEST gene (maternal methylation gene region, paternal methylation was preferentially expressed) were 0.66, 0.66 and 0.64, respectively, suggesting a possible low proportion mosaic UPD (7) mat. However, the experimental results are near the threshold range, so it cannot be interpreted accurately.
Twenty-one cases of fetal LOH were further verified by trio-WES, of which one (4.8%) had paternal UPD and four (19.0%) had maternal UPD. Notably, trio-WES was performed in Case 12, confirming maternal UPD15 associated with PWS, and the pregnancy was terminated at 28 weeks (Table 2). No definite imprinted genes were observed in the other four UPDs, and these were classified as VOUS.
Karyotyping results
Karyotyping was performed successfully in all 106 fetuses with LOH. In total, 97 cases yielded normal results, seven cases had abnormal karyotyping results, including 47,XX, + mar dn, 46,X,i(Xq)[40]/45,X[14], 46,XX,t(4;5)(q31;q23)pat, 46,XY,t(11;22)(q24.1;q12.3)pat, 46,XX,t(12;21)(q12;q22.2)mat, 47,XX, + 21, and mos 47,XY, + 22[3]/46,XY[58], and 2 cases had normal polymorphic variation (46, XY, 15ph, and 46,X,inv(Y)(p11.2q11.2) mos 45%, respectively).
Gene mutation results
Trio-WES was performed in 21 cases to detect gene mutation of autosomal recessive diseases in addition to UPD, and 11 results were clinically significant, including five pathogenic variants, four likely pathogenic variants, and two pathogenic UPDs. Among these clinically significant results, trio-WES identified homozygous mutations in autosomal recessive diseases attributed to LOHs in three cases (Cases 1, 9, and 15).
In Case 11, fetal BPD was small for gestational age as detected by ultrasound, SNP array showed a 38 Mb LOH in 14q13.1q24.2, prenatal trio-WES was declined, and the fetus was delivered at term. At five months old, the infant was 60 cm tall, and often arched her back; brain MRI at three months of age revealed asymmetrical bilateral ventricles, the left lateral ventricle was larger than the right, and some extracerebral spaces were slightly widened. Low T1W1 and high T2W1 signals were observed in the bilateral maxillary, ethmoid, and sphenoid sinuses. Postnatal trio-WES ruled out UPD14, and showed a de novo heterozygous mutation, NM_000095: c.1417_1419dup (p. D473dup), in COMP in the female infant (Fig. 3), which was an incidental finding associated with autosomal dominant pesudoachondroplasia (PSACH, OMIM:177,170), multiple epiphyseal dysplasia 1 (EDM1, OMIM:132,400), and carpal tunnel syndrome 2 (CTS2, OMIM:619,161). In Case 18, amniocentesis was performed, as the fetal left femur was slightly curved, and the SNP array revealed a 20 Mb LOH located in 5q23.2q32. Prenatal trio-WES indicated a de novo missense variant, NM_000088: c.1436G > C p.G479A , in COL1A1 (120,150) on chromosome 17 in the fetus (Fig. 4), associated with osteogenesis imperfecta, type I (OMIM:166,200), type II (OMIM:166,210), type III (OMIM:259,420), type IV (OMIM:166,220), Ehlers-Danlos syndrome, arthrochalasia type 1 (OMIM:130,060), Caffey disease (OMIM:114,000), and bone mineral density variation QTL, steoporosis (OMIM:166,710); the couple elected TOP at 26 weeks. In Case 29, amniocentesis was performed, as the fetal bilateral femoral curvature was less than the normal predictive value -2SD on ultrasound, and the SNP array revealed a 46.1 Mb LOH located in 5p15.33p11. Prenatal trio-WES indicated an inherited paternally splicing variant, NM_000088.4: c.1615-1G > T (p.G802V), in COL1A1 (120,150) on chromosome 17 in the fetus (Fig. 5), associated with autosomal dominant osteogenesis imperfecta, and the pregnancy was terminated at 23 weeks (Table 2).

Trio whole exome sequencing results for case 15. The female infant harbored a de novo heterozygous mutation, NM _000095: c.1417_1419dup(p.D473dup), in COMP in the female infant. (A) The female infant; (B) The mother; (C) The father.

Trio whole exome sequencing results for case 18. The fetus harbored a de novo missense variant, NM_000088: c.1436G > C p.G479A, in COL1A1 on chromosome 17. (A) The fetus; (B) The mother; (C) The father.

Trio whole-exome sequencing results for case 29. The fetus harbored an inherited paternally splicing variant, NM_000088.4: c.1615-1G > T (p. G802V), in COL1A1 on chromosome 17.
Perinatal outcome and follow-up
The pregnancy outcomes of the 106 fetuses with LOH were as follows: 67 term births (of which two resulted in neonatal death, and one had an abnormal phenotype after birth), 29 TOPs, three preterm births, two fetal deaths, and five miscarriages. A full-term infant (Case 14) was diagnosed with feeding difficulties after birth, and three months after delivery, the child's height and development were normal, except for low weight. A preterm infant (Case 2) was diagnosed postnatally with autism. One fetus (Case 11) showed fetal BPD that was small for gestation age on prenatal ultrasound screening and was delivered vaginally at term. The infant showed growth delay (60 cm tall at five months old), hypotonia, and often arched her back. The rate of adverse pregnancy outcomes in fetuses with LOH with UAs was 52.4% (22/42), and 32.8% (21/64) in those that did not show positive ultrasound findings (p < 0.05).
In 42 fetuses with LOH accepted further genetic analysis, the pregnancy outcomes were as follows: 24 term births (of which two resulted in neonatal death (Case 1 and 5), and three had abnormal phenotypes after birth (Case 11 was diagnosed with short stature, and often arches her back, Case 14 had feeding difficulties, and Case 34 presented with micrognathia), 18 TOPs, and one preterm birth (Case 2 had hand deformity and autism). In 64 fetuses with LOH declined further genetic analysis, the pregnancy outcomes were as follows: 42 term births (two had abnormal phenotypes after birth (Case 68 was diagnosed with hydrocephalus, abnormal fingers on both hands, learning disability, and Case 106 presented with epilepsy, mild mental retardation), 12 TOPs, five missed abortions, four preterm births, and one fetal demise in utero. The rate of the adverse pregnancy outcomes in fetal LOH accepted further genetic analysis was 54.8% (23/42), and 37.5% (24/64) in those declined further genetic analysis (p < 0.05).
Discussion
In our cohort, we investigated the clinical significance of fetal LOH as well as the correlation between fetal LOH and its clinical features. The detection rate of LOH meeting the reporting threshold in our study was 0.96% (106/11,062), slightly lower than the 0.97% (100/10,294) reported by Liu et al14., but significantly higher than the 0.43% (22/5063) reported by Liang et al15., the difference might be attributed to different microarray platforms, the size threshold of LOH reported, sample sizes and the type of the specimen. The threshold in our cohort was set according to that reported by Rehder et al9. and Hoppman et al16. In addition, none of the clinically significant LOHs occurring in chromosome X were reported due to lack of adverse family history associated with X-linked disorders. When fetal LOH occurred on a single chromosome, chromosomes 6 and 5 were the most commonly involved; whereas Liu et al14. showed that LOH was more likely to occur in chromosomes X, 2, and 16. The discordance may be due to the array platform and the reporting threshold of LOH studied.
Clinically significant imprinting disorders should be valued, especially for UPD involving imprinted chromosomes 6, 7, 11, 14, 15, and 2017,18. The clinical significance of UPD is closely associated with the affected imprinted region and genes in addition to parental origin15,19. Furthermore, it is unclear whether imprinting affects UPD 1620,21,22, since the outcomes of the carriers were variable, from normal growth to delayed growth22. Notably, in Cases 21 and 27, pregnancies with confirmed maternal UPD16 were terminated owing to UAs and abnormal genetic results.
In our cohort, 62.3% (66/106) of fetuses with LOH presented UAs, the most common UA was FGR or FGR combined with other indications (18/66 (27.3%)), and the most frequently observed ultrasound structure anomalies were cardiovascular system (6/66 (9.1%)), skeletal (4/66 (6.1%)), and genitourinary malformations (4/66 (6.1%)). Seventeen percent (18/106) of fetuses with LOH had FGR, UPDs 2, 7, 14, 15, and 16 were the underlying genetic causes of FGR17,23,24. The possible pathogenesis encompasses homozygous pathogenic variants in single gene diseases, imprinting effects, or confined placental mosaicism (CPM)25. Thus, UPD is one possible genetic factor resulting in FGR. Monitoring fetal growth via ultrasound is essential for the management of fetal LOH. Indicative prenatal ultrasound findings can be observed in patients with Beckwith-Wiedemann syndrome and SRS26,27. In our cohort, four cases (Cases 6, 25, 26, and 30) with LOH showed FGR or FGR combined with other indications, of which four UPDs were confirmed. The genetic causes underlying FGR were maternal UPD7 and UPD11 associated with SRS17, and TOP was elected owing to unfavorable outcomes. Patients with UPD14 showed multiple UAs, resulting in unfavorable outcomes28,29. Paternal UPD14 associated with KOS was confirmed in Case 4 with polyhydramnios (amniotic fluid index: 38.7 cm), and the pregnancy was terminated. Notably, the rate of adverse pregnancy outcomes in fetuses with LOH and UAs (36.4%) was higher than in those without UAs (15.0%) (p < 0.05). Our data demonstrate that regular ultrasound screening is essential to closely monitor the development of fetuses with LOH.
LOH also provides certain signs for investigating homozygous variants in autosomal recessive single gene diseases besides UPD and imprinting effects. In three cases in our cohort, pathogenic homozygous variants in single-gene diseases were further identified via trio-WES, resulting in UAs. Therefore, trio-WES should be first performed for its ability to verify UPD as well as identify homozygous mutations simultaneously (Cases 11, 16, and 17). Furthermore, six cases (Cases 2, 11, 16, 17, 18, and 29) had incidental findings of clinically significant variants. Our study also shows that trio-WES could identify incidental pathogenic mutations in addition to homozygous variants attributed to LOH. Therefore, trio-WES should be recommended first for fetal LOH, especially in fetuses with structural anomalies and/or consanguineous parents.
The study had some limitations. First, although it was a retrospective multicenter study, the sample size was not large enough and the follow-up period was not long enough, so some clinical features may have been missed. Studies with larger populations and longer follow-up will be needed. Second, the parental origin of LOH was further identified in only 39.6% of cases and none of the placental tissue in these cases was further investigated to confirm CPM.
Conclusion
We explored the clinical significance and features of fetal LOH. Various molecular genetic testing techniques, such as parental SNP array verification, trio-WES, MS-MLPA, regular and systematic ultrasonic examination, and placental study when necessary, should be comprehensively performed to precisely assess the prognosis of fetal LOH and guide the management of the affected pregnancy.
Data availability
The data used to support the findings of this study are available from the corresponding author upon request.
Abbreviations
- BPD:
-
Biparietal diameter
- CMA:
-
Chromosomal microarray analysis
- CNV:
-
Copy number variation
- FL:
-
Femur length
- FGR:
-
Fetal growth restriction
- HC:
-
Head circumference
- IBD:
-
Identity by descent
- LOH:
-
Loss of heterozygosity
- MS-MLPA:
-
Methylation-specific multiplex ligation-dependent probe amplification
- NT:
-
Nuchal translucency
- PWS:
-
Prader–Willi syndrome
- KOS:
-
Kagami-Ogata syndrome
- SRS:
-
Silver-Russell syndrome
- TOP:
-
Termination of pregnancy
- UAs:
-
Ultrasound anomalies
- UPD:
-
Uniparental disomy
- WES:
-
Whole-exome sequencing
- VOUS:
-
Variants of uncertain significance
References
Levy, B. et al. Genomic imbalance in products of conception: single-nucleotide polymorphism chromosomal microarray analysis. Obstet. Gynecol. 124, 202–209. https://doi.org/10.1097/aog.0000000000000325 (2014).
Sahoo, T. et al. Comprehensive genetic analysis of pregnancy loss by chromosomal microarrays: Outcomes, benefits, and challenges. Genetics Med. Off. J. Am. Coll. Med. Genetics 19, 83–89. https://doi.org/10.1038/gim.2016.69 (2017).
Del Gaudio, D. et al. Diagnostic testing for uniparental disomy: A points to consider statement from the american college of medical genetics and genomics (ACMG). Genetics Med. Off. J. Am. Coll. Med. Genetics 22, 1133–1141. https://doi.org/10.1038/s41436-020-0782-9 (2020).
Ledbetter, D. H. & Engel, E. Uniparental disomy in humans: Development of an imprinting map and its implications for prenatal diagnosis. Human Mol. Genetics 4, 1757–1764. https://doi.org/10.1093/hmg/4.suppl_1.1757 (1995).
Miron, P. M. Preparation, culture, and analysis of amniotic fluid samples. Curr. Protoc. Hum. Genetics https://doi.org/10.1002/0471142905.hg0804s74 (2012).
Hastings, R., McGowan-Jordan, J. & Moore, S. Addenda to ISCN 2020. Cytogenet. Genome Res. 163, 1–4. https://doi.org/10.1159/000533170 (2023).
Xue, H. et al. Detection of copy number variation associated with ventriculomegaly in fetuses using single nucleotide polymorphism arrays. Sci. Rep. 11, 5291. https://doi.org/10.1038/s41598-021-83147-7 (2021).
Kearney, H. M., Thorland, E. C., Brown, K. K., Quintero-Rivera, F. & South, S. T. American college of medical genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genetics Med. Off. J. Am. Coll. Med. Genetics 13, 680–685. https://doi.org/10.1097/GIM.0b013e3182217a3a (2011).
Rehder, C. W. et al. American College of Medical Genetics and Genomics: Standards and guidelines for documenting suspected consanguinity as an incidental finding of genomic testing. Genetics Med. Off. J. Am. Coll. Med. Genetics 15, 150–152. https://doi.org/10.1038/gim.2012.169 (2013).
Gonzales, P. R. et al. Interpretation and reporting of large regions of homozygosity and suspected consanguinity/uniparental disomy, 2021 revision: A technical standard of the american college of medical genetics and genomics (ACMG). Genetics Med. Off. J. Am. Coll. Med. Genetics 24, 255–261. https://doi.org/10.1016/j.gim.2021.10.004 (2022).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinform. (Oxford, England) 25, 1754–1760. https://doi.org/10.1093/bioinformatics/btp324 (2009).
Tavtigian, S. V. et al. Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genetics Med. Off. J. Am. Coll. Med. Genetics 20, 1054–1060. https://doi.org/10.1038/gim.2017.210 (2018).
Vasconcelos, S., Ramalho, C., Marques, C. J. & Doria, S. Altered expression of epigenetic regulators and imprinted genes in human placenta and fetal tissues from second trimester spontaneous pregnancy losses. Epigenetics 14, 1234–1244. https://doi.org/10.1080/15592294.2019.1634988 (2019).
Liu, J. et al. Absence of heterozygosity detected by single-nucleotide polymorphism array in prenatal diagnosis. Ultrasound Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 57, 314–323. https://doi.org/10.1002/uog.21951 (2021).
Liang, B. et al. Prenatal diagnosis of fetuses with region of homozygosity detected by single nucleotide polymorphism array: A retrospective cohort study. J. Hum. Genetics 67, 629–638. https://doi.org/10.1038/s10038-022-01062-9 (2022).
Hoppman, N., Rumilla, K., Lauer, E., Kearney, H. & Thorland, E. Patterns of homozygosity in patients with uniparental disomy: Detection rate and suggested reporting thresholds for SNP microarrays. Genetics Med. Off. J. Am. Coll. Med. Genetics 20, 1522–1527. https://doi.org/10.1038/gim.2018.24 (2018).
Kotzot, D. Prenatal testing for uniparental disomy: Indications and clinical relevance. Ultrasound Obstet. Gynecol. Off. J. Int. Soc. Ultrasound Obstet. Gynecol. 31, 100–105. https://doi.org/10.1002/uog.5133 (2008).
Eggermann, T., Soellner, L., Buiting, K. & Kotzot, D. Mosaicism and uniparental disomy in prenatal diagnosis. Trends Mol. Med. 21, 77–87. https://doi.org/10.1016/j.molmed.2014.11.010 (2015).
Robinson, W. P. Mechanisms leading to uniparental disomy and their clinical consequences. BioEssays News Rev. Mol. Cell. Dev. Biol. 22, 452–459. https://doi.org/10.1002/(sici)1521-1878(200005)22:5%3c452::Aid-bies7%3e3.0.Co;2-k (2000).
Hansen, W. F. et al. Maternal uniparental disomy of chromosome 2 and confined placental mosaicism for trisomy 2 in a fetus with intrauterine growth restriction, hypospadias, and oligohydramnios. Prenat. Diagnos. 17, 443–450. https://doi.org/10.1002/(sici)1097-0223(199705)17:5%3c443::aid-pd82%3e3.0.co;2-2 (1997).
Scheuvens, R. et al. Maternal uniparental disomy of chromosome 16 [upd(16)mat]: Clinical features are rather caused by (hidden) trisomy 16 mosaicism than by upd(16)mat itself. Clin. Genetics 92, 45–51. https://doi.org/10.1111/cge.12958 (2017).
Kotzot, D. & Utermann, G. Uniparental disomy (UPD) other than 15: Phenotypes and bibliography updated. Am. J. Med. GeneticsPart A 136, 287–305. https://doi.org/10.1002/ajmg.a.30483 (2005).
Yingjun, X. et al. Chromosomal uniparental disomy 16 and fetal intrauterine growth restriction. Eur. J. Obstet. Gynecol. Reproduct. Biol. 211, 1–7. https://doi.org/10.1016/j.ejogrb.2016.12.019 (2017).
Carmichael, H., Shen, Y., Nguyen, T. T., Hirschhorn, J. N. & Dauber, A. Whole exome sequencing in a patient with uniparental disomy of chromosome 2 and a complex phenotype. Clin. Genetics 84, 213–222. https://doi.org/10.1111/cge.12064 (2013).
Wilkins-Haug, L., Quade, B. & Morton, C. C. Confined placental mosaicism as a risk factor among newborns with fetal growth restriction. Prenat. Diagnos. 26, 428–432. https://doi.org/10.1002/pd.1430 (2006).
Bruce, S. et al. Submicroscopic genomic alterations in Silver-Russell syndrome and Silver-Russell-like patients. J. Med. Genetics 47, 816–822. https://doi.org/10.1136/jmg.2009.069427 (2010).
Eggermann, T. et al. Prenatal molecular testing for Beckwith-Wiedemann and Silver-Russell syndromes: A challenge for molecular analysis and genetic counseling. Eur. J. Hum. Genetics EJHG 24, 784–793. https://doi.org/10.1038/ejhg.2015.224 (2016).
Watanabe, T. et al. Prenatal findings and epimutations for paternal uniparental disomy for chromosome 14 syndrome. The J. Obstet. Gynaecol. Res. 41, 1133–1136. https://doi.org/10.1111/jog.12665 (2015).
Chen, C. L. et al. Prenatal diagnosis of paternal uniparental disomy for chromosome 14 using a single-nucleotide-polymorphism-based microarray analysis: A case report. J. Formosan Med. Assoc. Taiwan yizhi 118, 739–742. https://doi.org/10.1016/j.jfma.2018.12.010 (2019).
Acknowledgements
We thank the family members who participated in our study. We also appreciate the obstetricians, radiographers, sonographer, and pediatricians who assisted our study.
Funding
This study was sponsored by the Joint Funds for the Innovation of Science and Technology, Fujian Province (no.2020Y9149), 2021 Fujian provincial health technology project (no.2021GGA051), and Natural Science Foundation of Fujian Province (no.2022J01421).
Author information
Authors and Affiliations
Contributions
H.X., L.Z. and A.Y. wrote the main manuscript text and X.C., L.X., M.L., N.L., Q.G., L.C., and H.H. prepared figures. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xue, H., Yu, A., Zhang, L. et al. Genetic testing for fetal loss of heterozygosity using single nucleotide polymorphism array and whole-exome sequencing. Sci Rep 14, 2190 (2024). https://doi.org/10.1038/s41598-024-52812-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-52812-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
