
Abstract
Magnetic anisotropy is crucial in examining suitable materials for magnetic functionalities because it affects their magnetic characteristics. In this study, disordered-perovskite RCr0.5Fe0.5O3 (R = Gd, Er) single crystals were synthesized and the influence of magnetic anisotropy and additional ordering of rare-earth moments on cryogenic magnetocaloric properties was investigated. Both GdCr0.5Fe0.5O3 (GCFO) and ErCr0.5Fe0.5O3 (ECFO) crystallize in an orthorhombic Pbnm structure with randomly distributed Cr3+ and Fe3+ ions. In GCFO, the long-range order of Gd3+ moments emerges at a temperature of TGd (the ordering temperature of Gd3+ moments) = 12 K. The relatively isotropic nature of large Gd3+ moment originating from zero orbital angular momentum exhibits giant and virtually isotropic magnetocaloric effect (MCE), with a maximum magnetic entropy change of \(\Delta {S}_{M}\) ≈ 50.0 J/kg·K. In ECFO, the highly anisotropic magnetizations result in a large rotating MCE characterized by a rotating magnetic entropy change \(\Delta {S}_{\theta }\) = 20.8 J/kg·K. These results indicate that a detailed understanding of magnetically anisotropic characteristics is the key for exploring improved functional properties in disordered perovskite oxides.
Similar content being viewed by others


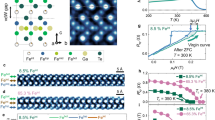
Introduction
Increased popularity of energy-efficient magnetic refrigeration in clean technology has inspired extensive research on novel magnetic materials to discover an effective technique of enhancing the magnetocaloric effect (MCE), which is described as the variation of temperature (T) in a magnetic material by applying the magnetic field (H)1,2,3,4. The MCE can be estimated by an adiabatic T change (\(\Delta {T}_{\mathrm{ad}}\)) and an isothermal magnetic entropy change (\(\Delta {\mathrm{S}}_{\mathrm{M}}\)) under the influence of H. Cryogenic magnetic refrigeration is crucial for obtaining sub-Kelvin temperatures as a substitute for3He/4He dilution refrigeration despite of increased cost and hydrogen gas liquefaction, which is utilized as an alternative fuel. Recently, large cryogenic MCE has been discovered in various insulating transition-metal oxides5,6,7 that possess easy manufacturability, chemical stability, and avoidance of refrigeration inefficiency because of eddy current. The beneficial aspect of MCE has been attained by the \(\Delta {T}_{\mathrm{ad}}\) in various oxide magnets, such as Gd2CoMnO68 (\(\Delta {T}_{\mathrm{ad}}\) = 1.3 K for ΔH = 0–9 T at 2 K and \(\Delta {T}_{\mathrm{ad}}\) = 8.3 K for ΔH = 0–9 T at 17 K ), SrFe0.5Co0.5O39 (\(\Delta {T}_{\mathrm{ad}}\) = 1.8 K for ΔH = 0–5 T at 330 K), HoMnO310 (\(\Delta {T}_{\mathrm{ad}}\) = 10.8 K for ΔH = 0–7 T at 11 K), CrO211 (\(\Delta {T}_{\mathrm{ad}}\) = 2.0 K for ΔH = 0–1.5 T at 390 K). Alternatively, the feasibility of magnetic refrigeration can be improved by developing a rotating MCE8,12,13, which can be achieved by rotating the refrigerant at constant H. The advantages of this method are technical simplicity and device compactness. However, strong magnetic anisotropy is essential for the realization of refrigerant cooling, which can be achieved using single-crystalline magnets whose intrinsic magnetocrystalline anisotropy originates from the anisotropic spin–orbit interaction that varies with the symmetry and structure. Cryogenic rotating MCE has been observed in several insulating oxide magnets, such as TbMnO314 (the magnetic entropy change obtained by rotation, \(\Delta {S}_{\theta }\) = 9.0 J/kg·K for 5 T at 15 K), HoMn2O515 (\(\Delta {S}_{\theta }\) = 12.4 J/kg·K for 7 T at 10 K), TmFeO316 (\(\Delta {S}_{\theta }\) = 9.0 J/kg·K for 5 T at 17 K), KTm(MoO4)217 (\(\Delta {S}_{\theta }\) = 9.8 J/kg·K for 5 T at 10 K), and KEr(MoO4)218 (\(\Delta {S}_{\theta }\) = 13 J/kg·K for 5 T at 10 K).
RCr0.5Fe0.5O3 (R = La, …, Lu) compounds crystallize in a disordered orthorhombic-perovskite structure with Pbnm space group having randomly distributed Cr3+ and Fe3+ ions owing to similar ionic radii of Cr3+ (0.615 Å) and Fe3+ (0.645 Å) ions19,20,21,22,23. The canted-antiferromagnetic order emerges in RFeO3 due to the dominant Fe3+-Fe3+ exchange couplings (Γ4(GxAyFz) in Bertaut’s notation)24. In RCr0.5Fe0.5O3, the Γ4 magnetic structure occurs at much lower T originating from a magnetic dilution effect of Cr3+ ions28. Extensive investigations on the series of compounds based on diverse magnetic phases and interactions reveal intriguing physical properties, such as metamagnetism25,26, exchange bias27,28, magnetodielectric effect22,29,30, and multiferroicity31,32,33,34. Additionally, large cryogenic MCEs in polycrystalline forms, such as GdCr0.5Fe0.5O320 (\(\Delta {S}_{M}\)= 29.2 J/kg·K for ΔH = 0–4.5 T), Gd2NiMnO635 (\(\Delta {S}_{M}\)= 37.2 J/kg·K for ΔH = 0–8 T), ErCr0.5Fe0.5O336 (\(\Delta {S}_{M}\)= 12.4 J/kg·K for ΔH = 0–5 T), and DyCr0.5Fe0.5O333 (\(\Delta {S}_{M}\)= 11.3 J/kg·K for ΔH = 0–4.5 T) have also been discovered. Various studies have hypothesized that large magnetic moments of magnetic rare-earth ions with strong anisotropy would significantly affect the cryogenic MCE. However, these studies focused only on polycrystalline specimens containing a large number of grains of all spatial orientations resulting in the average effect for observed physical properties.
To investigate the role of magnetic rare-earth ions and influence of anisotropic characteristics on MCE, single crystals of GdCr0.5Fe0.5O3 (GCFO) and ErCr0.5Fe0.5O3 (ECFO) were grown. For GCFO, large Gd3+ moments align below TGd (the ordering temperature of Gd3+ moments) = 12 K, which exhibits a relatively isotropic nature. The giant MCE is evidenced by the near-reversible magnetization along and perpendicular to the c-axis induces the maximum magnetic entropy change of \(\Delta {S}_{M}\) = 49.8 and 48.8 J/kg·K, respectively. In contrast, Er3+ moments aligned along the c-axis below TEr (the ordering temperature of Er3+ moments) = 11 K give rise to a highly anisotropic MCE. This generates a giant rotational MCE, i.e., \(\Delta {S}_{\theta }\) = 20.8 J/kg·K. In view of the distinct magnetic aspects of disordered perovskites, these results contribute to the fundamental and applied research on magnetic materials.
Results and discussion
Figure 1a and 1b show the X-ray diffraction patterns measured at room temperature for the ground GCFO and ECFO, and the simulated patterns analyzed by the Rietveld refinement using the Fullprof software, respectively. The refined results indicate that GCFO and ECFO form an orthorhombic disordered perovskite with the Pbnm space group. The lattice constants were observed to be a = 5.3318 Å, b = 5.5674 Å, and c = 7.6379 Å for GCFO and a = 5.2411 Å, b = 5.5451 Å, and c = 7.5496 Å for ECFO. Additional details of crystallographic data are summarized in Table 1. The crystallographic structures of GCFO and ECFO viewed from the c- and a-axes are depicted in Fig. 1c and 1d, respectively. These structures are distinct from a double perovskite in which two different transition metal ions are alternately located in corner-shared octahedral units. Therefore, in disordered perovskites GCFO and ECFO, the sites of Cr3+ and Fe3+ ions present the randomly distributed arrangement arising from comparable ionic radii. The oxygen octahedral cages are considerably distorted because of the small radius of Gd3+/Er3+ ions, resulting in O2- ion shifts in the bonds connecting the Cr3+/Fe3+ ions.

Crystallographic structures of GCFO and ECFO single crystals. (a, b) Observed (open circles) and calculated (solid line) powder X-ray diffraction patterns for the ground GdCr0.5Fe0.5O3 (GCFO) and ErCr0.5Fe0.5O3 (ECFO) single crystals. The short green lines denote the Bragg positions, and the blue curve indicates the difference between the observed and calculated patterns. (c) View of the crystallographic structure of GCFO from the c-axis and a-axis. The light blue, purple, and yellow spheres represent the Gd3+, Cr3+/Fe3+, and O2− ions, respectively. The black box with the rectangular cross-sections depict a crystallographic unit cell. (d) View of the crystallographic structure of ECFO from the c-axis and a-axis. The green, purple, and yellow spheres represent the Er3+, Cr3+/Fe3+, and O2- ions, respectively.
To examine the magnetic properties of GCFO and ECFO single crystals, the dependence of T on magnetic susceptibility \(\chi =\) M/H was measured at \(H\) = 0.01 T on warming after zero-field cooling (ZFC) and cooling in the same field (FC). The anisotropic \(\chi\)’s were obtained for the orientations that are parallel (H//c) and perpendicular to the c-axis (H \(\perp\) c), as shown in Fig. 2a and b for GCFO and Fig. 2c and d for ECFO, respectively. Based on a previous study, the canted antiferromagnetic order of Fe3+ magnetic moments in GdFeO3 manifests at TN = 661 K37. Unlike other orthoferrites, the canted moments along the c-axis do not rotate on further cooling. In GCFO, half of the Fe3+ ions are replaced by Cr3+ ions. However, the same tendency of canted moments aligned along the c-axis is sustained because \(\chi\) for H//c appears to be larger than that for H \(\perp\) c in the overall T range, except for the low-T regime (Fig. 2a and b). The Gd3+ moments in GCFO are antiferromagnetically ordered along magnetic easy-axis c in the low-T region, evidenced by the smaller magnitude and peaky feature of \(\chi\) for H//c. Even without an orbital moment (L = 0 of Gd), Gd compounds still possess a very small magnetic anisotropy due to weak dipole–dipole interaction of the large Gd spin. This weak anisotropy is the reason why the Gd spins are pointing towards the c-axis, i.e., a particular easy-axis38.

Magnetic susceptibility and heat capacity. (a, b) Temperature (T) dependence of magnetic susceptibility χ = M/H of single crystalline GCFO at H = 0.01 T measured on heating from 2 to 300 K after zero-field cooling (ZFC) and upon cooling at the same field (FC) parallel (H//c) and perpendicular (H \(\perp\) c) to the c-axis. (c, d) T dependence of magnetic susceptibility χ = M/H of single crystalline ECFO at H = 0.01 T measured on heating from 2 to 300 K after ZFC and FC for H//c and H \(\perp\) c. (e) Heat capacity divided by temperature (C/T) measured at H = 0 T and T = 2–300 K for GCFO. Inset shows C/T in the T = 2–35 K region. The gray curve was obtained by fitting, considering the influence of Cr3+/Fe3+ moments on C/T at a low-T regime. The vertical dashed line denotes the ordering T of Gd3+ moments as TGd = 12 K. The colored area indicates the contribution of Gd3+ ions to the magnetic entropy. (f) C/T measured at H = 0 T and T = 2–300 K for ECFO. Inset shows C/T in the T = 2–20 K region. The gray curve was obtained by fitting, considering the influence of Cr3+/Fe3+ moments on C/T at a low-T regime. The vertical dashed lines designate the ordering T of Er3+ moments as TEr = 11 K and the 2nd spin-reorientation transition of Cr3+/Fe3+ moments, TSR,2 = 7 K. The colored area indicates the contribution of Er3+ ions to the magnetic entropy.
In ErFeO3, the canted antiferromagnetic order of Fe3+ magnetic moments with a small net moment aligned along the c-axis occurs at TN ≈ 640 K39,40 with Γ4 magnetic structure41. On further cooling, the net magnetic moment rotates to the a-axis by 90˚ at TSR = 113 K by forming the Γ2(FxCyGz) magnetic structure, followed by the long range antiferromagnetic order of Er3+ magnetic moments aligned along the c-axis (Γ1(Cz-type order)) at T = 3.4 K42,43. In ECFO, the χ properties of H//c and H \(\perp\) c directions in the overall T range are strikingly different because of the strong anisotropic nature of the system (Fig. 2c and d). In contrast to the GCFO, the ECFO exhibits spin reorientation transition at TSR,1 ≈ 180 K, which indicates a considerable decrease in χ along the c-axis and an escalation of χ perpendicular to the c-axis. As shown in Fig. 3, we constructed the Belov-Arrott plot to determine the order of magnetic phase transition at TSR,1 ≈ 180 K. The slope was found to be positive for the overall regime of spin-reorientation, which suggests a second-order phase transition44,45. However, EFCO also exhibits signatures of a first-order phase transition such as thermally hysteretic behavior between ZFC and FC data (Fig. 2c) and the absence of a distinct peak in the specific heat (Fig. 2f). Thus, further studies are required to clearly identify the order of this spin-reorientation transition46,47. As T was further decreased, sharp anomalies were observed around 10 K indicating the antiferromagnetic order of Er3+ moments.

Belov-Arrott plot. Belov-Arrot plot for the ECFO crystal at H//c and T = 60—200 K.
The T dependence of the heat capacity divided by T (C/T) measured at H = 0 T for GCFO exhibits a sharp increase below TGd = 12 K, indicating the ordering of Gd3+ moments, as shown in Fig. 2e. The influence of the ordering of magnetic Gd3+ moments on C/T in the low T regime was estimated by subtracting the contributions from Cr3+ and Fe3+ ions below TGd. The subtracted part of C/T was obtained from the following equation:
where \(\gamma\), \(\rho\), \(\mathrm{and }\beta\) are coefficients corresponding to the electron, magnon, and phonon contributions of the Cr3+ and Fe3+ moments, respectively. Fitting the data to Eq. (1) resulted in the grey curve of C/T in the inset of Fig. 2e, which indicates the contribution from the interactions of Fe3+–Fe3+, Cr3+–Cr3+ and Cr3+–Fe3+ pairs and the interaction between the Gd3+ and Cr3+/Fe3+ sublattices at low T. The estimated entropy change based solely on the order of Gd3+ moments \(\Delta\) SGd in zero H was observed to be 7.5 J/mole∙K. \(\Delta\) SGd is 21.7% of the expected value of fully-saturated Gd3+ moments, i.e., \(2R\mathrm{ln}(2J+1)\) = 34.6 J/mole∙K, where R is the gas constant and \(J\) is the total angular momentum (\(J\) = 7/2 for Gd3+ ions).
Previous experimental studies on neutron diffraction on the polycrystalline ECFO suggest that the spin configuration transforms from representation Γ4(GxAyFz) to Γ2(FxCyGz) on lowering T across TSR,126. During the ordering of Er3+ moments at TEr = 11 K, the Cz component belonging to Γ1 was observed on the Er3+ sublattice. Further decrease in T causes the 2nd spin-reorientation transition at TSR,2 = 7 K on the Cr3+/Fe3+ sublattice where the Gy component identified as another Γ1 phase. Across TSR,2, larger Er3+ moments were also observed. Furthermore, the measured C/T value reveals two different transitions, i.e., TEr and TSR,2 at low-T regime, as shown in the inset of Fig. 2f. After subtracting the contribution of the Cr3+/Fe3+ sublattice represented by the gray curve, \(\Delta\) SEr in zero H was estimated to be 4.11 J/mole∙K, which is 8.9% of the expected value of the fully saturated Er3+ moments, \(2R\mathrm{ln}\left(2J+1\right)\) = 46.1 J/mole∙K (\(J\) = 15/2 for the Er3+ ions).
As shown in Fig. 4, the magnetic anisotropies in GCFO and ECFO were investigated using isothermal M for two different orientations (H//c and H \(\perp\) c) at 2 K, the former exhibiting an insignificant magnetic anisotropy. The initial M curve at H//c shows a weak magnetic transition at H = 0.85 T (Fig. 4a), suggesting a spin-flop transition because of the Gz component that is consistent with the transition observed in a previous study of the polycrystalline ECFO26. The value of M at a maximum H of 9 T is 6.75 μB/f.u. The consecutive sweeping of H reveals no hysteretic behavior with nearly zero remanent M and coercive field. The initial M curve at H \(\perp\) c increases smoothly (Fig. 4b) and attains the same magnitude of M at 9 T as that at H//c. Contrarily, ECFO has a distinctive H dependence for each direction. The slope of the isothermal M curve for H//c is the greatest in the narrow H regime between 0.3 and 1.0 T; thereafter, it decreases and the M value is observed to be 7.32 \({\mu }_{\mathrm{B}}\)/f.u. at 9 T. However, the initial M curve for H \(\perp\) c varies smoothly and does not reach saturation at H up to 9 T. The M value at 9 T is found to be 3.58 \({\mu }_{\mathrm{B}}\)/f.u., which is approximately half the value of M at 9 T for H//c.

Isothermal magnetization. (a, b) Full magnetic hysteresis curve of the isothermal magnetization measured at 2 K up to H = ± 9 T at H//c and H \(\perp\) c, respectively, for GCFO. (c, d) Full magnetic hysteresis curve of the isothermal magnetization measured at 2 K up to H = ± 9 T at H//c and H \(\perp\) c, respectively, for GCFO.
In GCFO, same M values at the maximum H and similar shapes of M curves for different orientations imply the moderately isotropic nature of the Gd3+ moments associated with the half-filled 4f. electronic configuration (S = 7/2 and L = 0). Therefore, the crystal field effect affected by the symmetry of local environment can be minimum48,49,50. Contrarily, the Er3+ ion exhibits strong anisotropic properties in the ECFO system because its angular momentum (L = 6) breaks the local symmetry and the crystal field effect substantially affects the magnetocrystalline anisotropy43,50.
The contrasting magnetic properties of GCFO and ECFO lead to different MCE characteristics measured using the initial M curves with dense T steps for T = 2–30 K, as shown in Fig. 5. In GCFO, the almost isotropic magnetic properties resulted in the typical decreasing trend of the M values similarly for the two different orientations as T is increased (Fig. 5a and 5b). For H//c in ECFO, rapid increase in the initial M curve in the low-H regime at 2 K becomes broaden as T increases; hence, the M value at low H is lower than that at higher T, as plotted in the inset of Fig. 5c. This characteristic of the initial M curves varies above 10 K; thus, the M value exhibits a typical reduction in most of the H regime as T increases. At H \(\perp\) c, owing to the smaller magnitude and smooth variation of M values, the overall magnitude of M is reduced marginally but continually in the entire regime of H as T increases (Fig. 5d).

Initial curves of isothermal magnetization. (a, b) Initial curves of the isothermal magnetization for H//c and H \(\perp\) c, respectively, at temperatures varying from 2 to 30 K for GCFO. (c, d) Initial curves of the isothermal magnetization for H//c and H \(\perp\) c, respectively, at temperatures varying from 2 to 30 K for ECFO. Inset of c shows the initial curves of magnetization in the low-H region for T = 2, 4, 6, 8, 10, 15, and 25 K.
The MCE in GCFO and ECFO was quantified by calculating the isothermal magnetic entropy change, \(\Delta {S}_{M}\), at a given T using the Maxwell’s relation:
where \({\mu }_{o}\) is the magnetic permeability of free space, Hf is the end point of H for the integral (Hf = 3, 5, 7, and 9 T), and the T gradient of M. \(\frac{\partial M\left(T,H\right)}{\partial T}\) was estimated using the slope of two consecutive data points. The T-dependence of estimated \(\Delta {S}_{M}(T)\) for H//c and H \(\perp\) c is plotted in Fig. 6, for the H regimes of ΔH = 0–3, 0–5, 0–7, and 0–9 T respectively. The \(\Delta {S}_{M}\) values for both orientations in GCFO present the largest at 4 K, where the maximum \(\Delta {S}_{M}\) for ΔH = 0–9 T were achieved as 49.8 and 48.8 J/kg·K, respectively, for H//c and H \(\perp\) c (Fig. 6a and b). This value of \(\Delta {S}_{M}\) is greater than that of the other oxide materials, such as Dy2CoMnO651 (\(\Delta {S}_{M}\)= 9.3 J/kg·K for ΔH = 0–7 T), HoMnO352 (\(\Delta {S}_{M}\)= 13.1 J/kg·K for ΔH = 0–7 T), GdCrO453 (\(\Delta {S}_{M}\)= 29.0 J/kg·K for ΔH = 0–9 T), and HoCrO454 (\(\Delta {S}_{M}\)= 31.0 J/kg·K for ΔH = 0–8 T). Furthermore, non-hysteretic behavior of isothermal M indicates the absence of unnecessary \(\Delta {S}_{M}\) loss.

Anisotropic magnetocaloric effect in GCFO and ECFO crystals. (a, b) Anisotropic magnetocaloric effect in GCFO. T dependence of magnetic entropy change \(-\Delta {S}_{M}\) for H//c and H \(\perp\) c with H regimes of \(\Delta H=\) 0–3, 0–5, 0–7, and 0–9 T, obtained by integrating the T gradient of the initial magnetization curves in Fig. 4a and b, respectively. (c, d) Anisotropic magnetocaloric effect in ECFO. T dependence of magnetic entropy change \(-\Delta {S}_{M}\) for H//c and H \(\perp\) c with H regimes of \(\Delta H=\) 0–3, 0–5, 0–7, and 0–9 T, obtained by integrating the T gradient of the initial magnetization curves in Fig. 4c and d, respectively.
In ECFO, the peculiar anisotropy of \(\Delta {S}_{M}\) was observed because the Er3+ spins mainly aligned along the c-axis. The intercrossed isothermal M values in the low-H regime for H//c (inset of Fig. 5c) considerably cancelled \(\Delta {S}_{M}\); hence, \(\Delta {S}_{M}\) for ΔH = 0–9 T was calculated to be 6.5 J/kg·K at 3 K (Fig. 6c). As T increases further, \(\Delta {S}_{M}\) continues to increase and peaks sharply at TSR,2 with a maximum \(\Delta {S}_{M}\) value of 39.1 J/kg·K. This feature was derived from the largest drop of isothermal M across TSR,2, which was verified by measuring initial M curves repeatedly in the low-T regime for various samples of ECFO crystals. Above TSR,2, \(\Delta {S}_{M}\) shows a broad variation and its value is approximately 20 J/kg·K. In contrast with \(\Delta {S}_{M}\) for H//c, the overall magnitude of \(\Delta {S}_{M}\) for H \(\perp\) c is largely reduced and its maximum value turns out to be 13.2 J/kg·K for ΔH = 0–9 T (Fig. 6d). Additionally, \(\Delta {S}_{M}\) was estimated up to T = 200 K for investigating the influence of χ variation at H = 0.01 T across the spin-reorientation transition of TSR,1 (Figs. 2c and d). As shown in Fig. 7, magnitude and anisotropy of the estimated \(\Delta {S}_{M}\) relevant to the spin-reorientation of Cr3+/Fe3+ moments were not pronounced. We also estimated relative cooling power (RCP) for both GCFO and ECFO crystals to show the potential of our single crystals as magnetic cryo-refrigerant. The RCP can be expressed by the following equation,
where Tcold = 2 K and Thot was determined by the full width half maximum in \(\Delta {S}_{M}\). Due to the rather isotropic nature of MCE, the RCP in the GCFO was estimated as 301 and 309 J/ kg at H//c and H \(\perp\) c, respectively, for ΔH = 0–9 T. On the other hand, the RCP in the ECFO was found to be 623 J/kg at H//c and 169 J/kg at H \(\perp\) c. The RCP has been estimated in polycrystalline specimens such as La0.67Sr0.22Ba0.11Mn0.9Fe0.1O355 (241 J/kg for ΔH = 0–5 T), La0.57Mg0.23MnO356 (176 J/kg for ΔH = 0–5 T), Ni0.5Zn0.5Fe2O457 (161 J/kg for ΔH = 0–2.5 T) and La0.5Pr0.2Ca0.1Sr0.2MnO358 (372 J/kg for ΔH = 0–5 T), and in single crystals such as La0.7Ca0.3MnO359 (358 J/kg for ΔH = 0–5 T), h-DyMnO360 (300 J/kg for ΔH = 0–5 T) and GdScO361 (307 J/kg) for ΔH = 0–7 T).

Anisotropic magnetic entropy change in ECFO. (a) T dependence of magnetic entropy change \(-\Delta {S}_{M}\) for H//c with magnetic field regimes of \(\Delta H=\) 0–3, 0–5, 0–7, and 0–9 T, respectively, obtained by integrating the T gradient of the initial magnetization curves in ECFO in the range T = 2–200 K. (b) T dependence of magnetic entropy change \(-\Delta {S}_{M}\) for H \(\perp\) c with magnetic field regimes of \(\Delta H=\) 0–3, 0–5, 0–7, and 0–9 T, respectively, obtained by integrating the T gradient of the initial magnetization curves in ECFO in the range T = 2–200 K.
To employ the conspicuous characteristics of the anisotropic MCE in disordered-perovskite ECFO, the rotating MCE was detected using the angle dependence of \(\Delta {S}_{M}\), which is denoted by \(\Delta {S}_{\theta }\), where \(\theta\) is the angle deviated from the c-axis (\(\theta\) = 0° for H//c, and \(\theta\) = 90° for H \(\perp\) c), as shown in the inset of Fig. 7. Figure 8 displays the resulting \(\Delta {S}_{\theta }\) taken at T = 3, 7, 10 and 29 K for H = 9 T. The dissimilar T dependence of \(\Delta {S}_{M}\) between H//c and H \(\perp\) c in the low T regime (Fig. 6c and d) engenders the angle-dependent modulation of \(\Delta {S}_{\theta }\), which changes significantly with T. \(\Delta {S}_{\theta }\) at 3 K alters negligibly with θ rotation; however, \(\Delta {S}_{\theta }\) at TSR,2 = 7 K shows a sudden increase to 15°. The continued variation in \(\Delta {S}_{\theta }\) with θ yields a large rotational MCE demonstrated by the maximum change of \(\Delta {S}_{\theta }\) = 20.8 J/kg·K, which would have applications in the rotary magnetic refrigerator technology. We have estimated the magnetic anisotropy constant for the ECFO crystal. The magnetic free energy of the ECFO crystal with uniaxial magnetocrystalline anisotropy can be described by \(F=K{\mathrm{sin}}^{2}\theta -MH\mathrm{cos}\theta ,\) where the first term indicates the magnetic anisotropy energy with the angle θ deviating from the c axis and the second term denotes the Zeeman energy. The magnetic anisotropy constant \(K\) was determined by the Sucksmith and Thompson method62, based on the experimental data for the ECFO crystal. The result shown in Fig. 9 exhibits a clear feature at TSR,2 = 7 K63. As T increases further, the gradual increase of \(\Delta {S}_{\theta }\) with θ demonstrates a maximum \(\Delta {S}_{\theta }\) of 9.9 and 13.5 J/kg·K at 10 and 29 K, respectively.

Rotating magnetocaloric effect in ECFO. Angular dependence of magnetic entropy change \(\Delta {S}_{\theta }\) at T = 3, 7, 10, and 29 K with \(\Delta H\) = 0–9 T, respectively. \(\theta\) is the angle deviating from the c-axis, i.e., \(\theta =0^\circ\) for H//c and \(90^\circ\) for H \(\perp\) c.

Magnetic anisotropy constant. Temperature dependence of magnetic anisotropy constant \(K\).
Conclusion
This study investigated the anisotropic magnetic and magnetocaloric properties of disordered-perovskite GdCr0.5Fe0.5O3 and ErCr0.5Fe0.5O3. In GdCr0.5Fe0.5O3, the limited isotropic nature of Gd3+ moments owing to zero orbital angular momentum creates a weak directional dependence of giant magnetocaloric effect, characterized by maximum magnetic entropy changes of \(\Delta {S}_{M}\) = 49.8 and 48.8 J/kg·K along and perpendicular to the c-axis, respectively. Non-magnetic hysteretic behavior of isothermal magnetization indicates the absence of dispensable loss of magnetocaloric effect. In contrast, ErCr0.5Fe0.5O3 reveals the sharpened feature of \(\Delta {S}_{M}\) that is derived from the largest reduction of isothermal magnetization between adjacent measuring temperatures across the 2nd spin-reorientation along the c-axis at TSR,2 = 7 K. This particular anisotropy causes large rotary magnetocaloric effect with a maximum entropy change of \(\Delta {S}_{\theta }\) = 20.8 J/kg·K. The results on different anisotropic magnetic properties of the disordered-perovskite compounds provide insights into suitable materials for magnetic functional applications.
Methods
Single crystals of GCFO and ECFO were synthesized using conventional flux method with PbO, PbO2, and PbF2 fluxes in a high-T furnace. The stoichiometric ratios of Gd2O3/Er2O3, Cr2O3, and Fe2O3 powders for GCFO and ECFO were mixed and ground using a pestle in a corundum mortar. The mixture was pelletized and calcined at 1000 °C for 12 h. The calcined pellet was finely re-ground, pelletized, and sintered at 1200 °C for 12 h. The same procedure was repeated at 1250 °C for 24 h. The pre-sintered power containing fluxes was heated to 1260 °C in a platinum crucible for 24 h until it was completely dissolved. Thereafter, it was slowly cooled to 850 °C at the rate of 2 °C/h and further cooled to room temperature T at the rate of 100 °C/h. To identify the crystallographic structures of GCFO and ECFO crystals, X-ray diffraction was performed using an X-ray diffractometer (Ultima IV, Rigaku Corp., Japan) with Cu-\({\mathrm{\rm K}}_{\mathrm{\alpha }}\) radiation.
The T and H dependences of DC magnetization (M) were obtained using a vibrating sample magnetometer at T = 2–300 K and H = -9–9 T in a physical properties measurement system (PPMS, Quantum Design, Inc., USA). The dependence of T on specific heat (C) was measured using the standard relaxation method and PPMS.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request. The datasets associated with the crystallographic structures that are analyzed in this study are available in the Crystallography Open Database (COD) repository, #3,000,434 (https://www.crystallography.net/cod/information_card.php?id=3000434&CODSESSION=cbg4dlc90k4ciiuucsm3fkq2a3) and #3,000,435 (https://www.crystallography.net/cod/information_card.php?id=3000435&CODSESSION=cbg4dlc90k4ciiuucsm3fkq2a3).
References
Pecharsky, V. K. & Gschneidner, K. A. Jr. Magnetocaloric effect and magnetic refrigeration. J. Magn. Magn. Mater. 200, 44–56. https://doi.org/10.1016/S0304-8853(99)00397-2 (1999).
de Oliveira, N. A. & von Ranke, P. J. Theoretical aspects of the magnetocaloric effect. Phys. Rep. 489, 89–159. https://doi.org/10.1016/j.physrep.2009.12.006 (2010).
Franco, V. et al. Magnetocaloric effect: From materials research to refrigeration devices. Prog. Mater. Sci. 93, 112–232. https://doi.org/10.1016/j.pmatsci.2017.10.005 (2018).
Evangelisti, M. et al. Cryogenic magnetocaloric effect in a ferromagnetic molecular dimer. Angew. Chem. Int. Ed. Engl. 50, 6606–6609. https://doi.org/10.1002/anie.201102640 (2011).
Phan, M.-H. & Yu, S.-C. Review of the magnetocaloric effect in manganite materials. J. Magn. Magn. Mater. 308, 325–340. https://doi.org/10.1016/j.jmmm.2006.07.025 (2007).
Ram, N. R. et al. Review on magnetocaloric effect and materials. J. Supercond. Nov. Magn. 31, 1971–1979. https://doi.org/10.1007/s10948-018-4666-z (2018).
Zhang, Y. et al. Magnetic properties and giant cryogenic magnetocaloric effect in B-site ordered antiferromagnetic Gd2MgTiO6 double perovskite oxide. Acta. Mater. 226, 117669. https://doi.org/10.1016/j.actamat.2022.117669 (2022).
Moon, J. Y., Kim, M. K., Choi, Y. J. & Lee, N. Giant anisotropic magnetocaloric effect in double-perovskite Gd2CoMnO6 single crystals. Sci. Rep. 7, 16099. https://doi.org/10.1038/s41598-017-16416-z (2017).
Yin, C. et al. Unusual oxidation states give reversible room temperature magnetocaloric effect on perovskite-related oxides SrFe0.5Co0.5O3. J. Solid State Chem. 184, 3228–3231. https://doi.org/10.1016/j.jssc.2011.10.002 (2011).
Balli, M. et al. Magnetocaloric properties of the hexagonal HoMnO3 single crystal revisited. Physica B Condens. 478, 77–83. https://doi.org/10.1016/j.physb.2015.08.063 (2015).
Zhang, X., Chen, Y., Lü, L. & Li, Z. A potential oxide for magnetic refrigeration application: CrO2 particles. J. Phys.: Condens. Matter 18, L559. https://doi.org/10.1088/0953-8984/18/44/L01 (2006).
Moon, J. Y. et al. Anisotropic magnetic properties and giant rotating magnetocaloric effect in double-perovskite Tb2CoMnO6. Phys. Rev. B https://doi.org/10.1103/PhysRevB.98.174424 (2018).
Nikitin, S. A., Skokov, K. P., Koshkidko, Y. S., Pastushenkov, Y. G. & Ivanova, T. I. Giant rotating magnetocaloric effect in the region of spin-reorientation transition in the NdCo5 single crystal. Phys. Rev. Lett. 105, 137205. https://doi.org/10.1103/PhysRevLett.105.137205 (2010).
Jin, J.-L. et al. Giant anisotropy of magnetocaloric effect in TbMnO3 single crystals. Phys. Rev. B 83, 184431. https://doi.org/10.1103/PhysRevB.83.184431 (2011).
Balli, M., Jandl, S., Fournier, P. & Gospodinov, M. M. Anisotropy-enhanced giant reversible rotating magnetocaloric effect in HoMn2O5 single crystals. Appl. Phys. Lett. 104, 232402. https://doi.org/10.1063/1.4880818 (2014).
Ke, Y.-J., Zhang, X.-Q., Ma, Y. & Cheng, Z.-H. Anisotropic magnetic entropy change in RFeO3 single crystals(R = Tb, Tm, or Y). Sci. Rep. 6, 19775. https://doi.org/10.1038/srep19775 (2016).
Tarasenko, R., Tkáč, V., Orendáčová, A., Orendáč, M. & Feher, A. Experimental study of the rotational magnetocaloric effect in KTm(MoO4)2. Phys. B Condens. 538, 116–119. https://doi.org/10.1016/j.physb.2018.03.027 (2018).
Tkáč, V. et al. Giant reversible rotating cryomagnetocaloric effect in KEr(MoO4)2 induced by a crystal-field anisotropy. Phys. Rev. B 92, 024406. https://doi.org/10.1103/PhysRevB.92.024406 (2015).
Pomiro, F. et al. Spin reorientation, magnetization reversal, and negative thermal expansion observed in RFe0.5Cr0.5O3 perovskites (R=Lu, Yb, Tm). Phys. Rev. B 94, 134402. https://doi.org/10.1103/PhysRevB.94.134402 (2016).
Yin, L. H. et al. Magnetocaloric effect and influence of Fe/Cr disorder on the magnetization reversal and dielectric relaxation in RFe0.5Cr0.5O3 systems. Appl. Phys. Lett. 110, 192904. https://doi.org/10.1063/1.4983363 (2017).
Yin, L. H. et al. Multiple temperature-induced magnetization reversals in SmCr1−xFexO3 system. Mater. Res. Bull. 48, 4016–4021. https://doi.org/10.1016/j.materresbull.2013.06.016 (2013).
Shin, H. J., Lee, N. & Choi, Y. J. Nonlinear magnetodielectric effect of disordered perovskite HoCr0.5Fe0.5O3: Role of magnetic rare-earth ions. J. Alloys Compd. 785, 1166–1172. https://doi.org/10.1016/j.jallcom.2019.01.291 (2019).
Mao, J. et al. Temperature- and magnetic-field-induced magnetization reversal in perovskite YFe0.5Cr0.5O3. Appl. Phys. Lett. 98, 192510. https://doi.org/10.1063/1.3590714 (2011).
Bertaut, E. F. in Spin Arrangements and Crystal Structure, Domains, and Micromagnetics (eds George T. Rado & Harry Suhl) 149–209 (Academic Press, 1963).
Nair, V. G., Pal, L., Subramanian, V. & Santhosh, P. N. Structural, magnetic, and magnetodielectric studies of metamagnetic DyFe0.5Cr0.5O3. J. Appl. Phys. 115, 17D728. https://doi.org/10.1063/1.4870139 (2014).
Bolletta, J. P. et al. Spin reorientation and metamagnetic transitions in RFe0.5Cr0.5O3 perovskites (R=Tb, Dy, Ho, Er). Phys. Rev. B 98, 134417. https://doi.org/10.1103/PhysRevB.98.134417 (2018).
Sharannia, M. P. et al. Observation of magnetization and exchange bias reversals in NdFe0.5Cr0.5O3. J. Magn. Magn. Mater. 430, 109–113. https://doi.org/10.1016/j.jmmm.2016.12.035 (2017).
Hou, L., Shi, L., Zhao, J., Tong, R. & Xin, Y. Insight into the magnetization reversal and exchange bias in RFe0.5Cr0.5O3 ceramics. J. Phys. Chem. C 125, 7950–7958. https://doi.org/10.1021/acs.jpcc.1c00562 (2021).
Mandal, P. et al. Spin-reorientation, ferroelectricity, and magnetodielectric effect in YFe1-xMnxO3 (0.1≤ x ≤ 0.40). Phys. Rev. Lett. 107, 137202. https://doi.org/10.1103/PhysRevLett.107.137202 (2011).
Kumar, A. et al. Origin of natural and magnetic field induced polar order in orthorhombic PrFe1/2Cr1/2O3. Phys. Rev. B 104, 035101. https://doi.org/10.1103/PhysRevB.104.035101 (2021).
Tokunaga, Y. et al. Composite domain walls in a multiferroic perovskite ferrite. Nat. Mater. 8, 558–562. https://doi.org/10.1038/nmat2469 (2009).
Das, N. et al. Pr2FeCrO6: A type I multiferroic. Inorg. Chem. 56, 12712–12718. https://doi.org/10.1021/acs.inorgchem.7b01086 (2017).
Yin, L. H. et al. Multiferroicity and magnetoelectric coupling enhanced large magnetocaloric effect in DyFe0.5Cr0.5O3. Appl. Phys. Lett. https://doi.org/10.1063/1.4862665 (2014).
Lohr, J. et al. Multiferroic properties of RFe0.5Cr0.5O3 with R=Tm, Er, Ho, Dy, and Tb. Phys. Rev. B 98, 134405. https://doi.org/10.1103/PhysRevB.98.134405 (2018).
Ali, A., Pasrija, K., Sharma, G., Kumar, S. & Singh, Y. Rare-earth tuned magnetism and magnetocaloric effects in double perovskites R2NiMnO6. J. Phys.: Condens. Matter 34, 095803. https://doi.org/10.1088/1361-648X/ac3e9e (2022).
Yadav, K., Kaur, G., Sharma, M. K. & Mukherjee, K. Magnetocaloric effect and spin-phonon correlations in RFe0.5Cr0.5O3 (R = Er and Yb) compounds. Phys. Lett. A 384, 126638. https://doi.org/10.1016/j.physleta.2020.126638 (2020).
Treves, D. Studies on orthoferrites at the Weizmann institute of science. J. Appl. Phys. 36, 1033–1039. https://doi.org/10.1063/1.1714088 (1965).
Rotter, M. et al. Dipole interaction and magnetic anisotropy in gadolinium compounds. Phys. Rev. B 68, 144418. https://doi.org/10.1103/PhysRevB.68.144418 (2003).
Eibschütz, M., Shtrikman, S. & Treves, D. Mössbauer studies of Fe57 in orthoferrites. Phys. Rev. 156, 562–577. https://doi.org/10.1103/PhysRev.156.562 (1967).
Zhou, Z., Guo, L., Yang, H., Liu, Q. & Ye, F. Hydrothermal synthesis and magnetic properties of multiferroic rare-earth orthoferrites. J. Alloys Compd. 583, 21–31. https://doi.org/10.1016/j.jallcom.2013.08.129 (2014).
Gorodetsky, G. et al. Magnetic structure of ErFeO3 below 4.5K. Phys. Rev. B 8, 3398–3404. https://doi.org/10.1103/PhysRevB.8.3398 (1973).
Sasani, A., Iñiguez, J. & Bousquet, E. Magnetic phase diagram of rare-earth orthorhombic perovskite oxides. Phys. Rev. B 104, 064431. https://doi.org/10.1103/PhysRevB.104.064431 (2021).
White, R. L. Review of recent work on the magnetic and spectroscopic properties of the rare-earth orthoferrites. J. Appl. Phys. 40, 1061–1069. https://doi.org/10.1063/1.1657530 (1969).
Gamża, M. B. et al. Complex magnetic phase diagram of metamagnetic MnPtSi. Phys. Rev. B 100, 014423. https://doi.org/10.1103/PhysRevB.100.014423 (2019).
Sharma, P. et al. Spin reorientation transition and spin dynamics study of perovskite orthoferrite TmFeO3 detected by electron paramagnetic resonance. Phys. Chem. Chem. Phys. 22, 21403–21411. https://doi.org/10.1039/D0CP00918K (2020).
Hou, L. et al. Spin-reorientation transition driven by double exchange in CeFeO3 ceramics. J. Phys. Chem. C 124, 15399–15405. https://doi.org/10.1021/acs.jpcc.0c00379 (2020).
Bonilla, C. M. et al. Universal behavior for magnetic entropy change in magnetocaloric materials: An analysis on the nature of phase transitions. Phys. Rev. B 81, 224424. https://doi.org/10.1103/PhysRevB.81.224424 (2010).
Gilleo, M. A. Magnetic properties of a gadolinium orthoferrite, GdFeO3. Crystal. J. Chem. Phys. 24, 1239–1243. https://doi.org/10.1063/1.1742747 (1956).
Gorodetsky, G. & Treves, D. Second-order susceptibility terms in orthoferrites at room temperature. Phys. Rev. 135, A97–A101. https://doi.org/10.1103/PhysRev.135.A97 (1964).
Mikami, I. Paramagnetic behavior of rare-earth ions in some orthoferrites. J. Phy. Soc. Jpn 34, 338–342. https://doi.org/10.1143/JPSJ.34.338 (1973).
Ganeshraj, C., Pradheesh, R. & Santhosh, P. N. Structural, magnetic, transport and magnetocaloric properties of metamagnetic DyMn0.5Co0.5O3. J. Appl. Phys. 111, 07A914. https://doi.org/10.1063/1.3672067 (2012).
Midya, A. et al. Magnetocaloric effect in HoMnO3 crystal. Appl. Phys. Lett. 96, 142514. https://doi.org/10.1063/1.3386541 (2010).
Palacios, E. et al. Effect of Gd polarization on the large magnetocaloric effect of GdCrO4 in a broad temperature range. Phys. Rev. B 93, 064420. https://doi.org/10.1103/PhysRevB.93.064420 (2016).
Midya, A., Khan, N., Bhoi, D. & Mandal, P. 3d–4f spin interaction induced giant magnetocaloric effect in zircon-type DyCrO4 and HoCrO4 compounds. Appl. Phys. Lett. 103, 092402. https://doi.org/10.1063/1.4819768 (2013).
Ben Hassine, R. et al. Enhanced relative cooling power of Fe-doped La0.67Sr0.22Ba0.11Mn1-xFexO3 perovskites: Structural, magnetic and magnetocaloric properties. J. Alloys Compd. 649, 996–1006. https://doi.org/10.1016/j.jallcom.2015.07.034 (2015).
Selmi, R., Chérif, W., Sarabando, A. R., Ferreira, N. M. & Ktari, L. Enhanced relative cooling power of lanthanum-deficiency manganites La0.77−xMg0.23MnO3 (0 ≤ x ≤ 0.2): Structural, magnetic and magnetocaloric properties. J. Mater. Sci.: Mater. Electron. 33, 1703–1723 (2022).
Anwar, M. S., Ahmed, F. & Koo, B. H. Enhanced relative cooling power of Ni1−xZnxFe2O4 (0.0⩽x⩽0.7) ferrites. Acta. Mater. 71, 100–107. https://doi.org/10.1016/j.actamat.2014.03.002 (2014).
Skini, R. et al. Large room temperature relative cooling power in La0.5Pr0.2Ca0.1Sr0.2MnO3. J. Alloys Compd. 827, 154292 (2020).
Debnath, J. C., Zeng, R., Kim, J. H., Chen, D. P. & Dou, S. X. Anisotropic and excellent magnetocaloric properties of La0.7Ca0.3MnO3 single crystal with anomalous magnetization. Mater. Sci. Eng. B 177, 48–53. https://doi.org/10.1016/j.mseb.2011.09.034 (2012).
Balli, M. et al. On the magnetocaloric effect in the multiferroic hexagonal DyMnO3 single crystals. J. Magn. Magn. Mater. 374, 252–257. https://doi.org/10.1016/j.jmmm.2014.08.009 (2015).
Jia, J.-H. et al. Giant magnetocaloric effect in the antiferromagnet GdScO3 single crystal. J. Alloys Compd. 803, 992–997. https://doi.org/10.1016/j.jallcom.2019.06.361 (2019).
Sucksmith, W. & Thompson, J. E. (1954) The magnetic anisotropy of cobalt. Proceedings of the Royal Society of London Series A. Mathematical and Physical Sciences 225, 362–375, https://doi.org/10.1098/rspa.1954.0209
Nikitin, S. A. et al. Magnetization, magnetic anisotropy and magnetocaloric effect of the Tb0.2Gd0.8 single crystal in high magnetic fields up to 1 T in region of a phase transition. Acta. Mater. 161, 331–337. https://doi.org/10.1016/j.actamat.2018.09.017 (2018).
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) through grants NRF-2017R1A5A1014862 (SRC program: vdWMRC center), NRF-2021R1A2C1006375, and NRF-2022R1A2C1006740. We would like to thank Editage (www.editage.co.kr) for English language editing.
Author information
Authors and Affiliations
Contributions
N.L. and Y.J.C. conceived the project. H.J.S. synthesized the single crystals. H.J.S., J.S.K, and J.H.K. measured the physical properties of the crystals. H.J.S., N.L. and Y.J.C. analyzed the data and prepared the manuscript. K.W.J. estimated the energy of magnetic anisotropy. All the authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shin, H.J., Kim, J.S., Jeong, K.W. et al. Giant and highly anisotropic magnetocaloric effects in single crystals of disordered-perovskite RCr0.5Fe0.5O3 (R = Gd, Er). Sci Rep 13, 7105 (2023). https://doi.org/10.1038/s41598-023-34258-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-34258-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
