
Abstract
In recent years, postmortem brain studies have revealed that some molecular, cellular, and circuit changes associated with suicide, have an independent or additive effect on depression. The aim of the present study is to identify potential phenotypic, tissue, and sex-specific novel targets and pathways to distinguish depression or suicide from major depressive disorder (MDD) comorbid with suicide. The mRNA expression profiling datasets from two previous independent postmortem brain studies of suicide and depression (GSE102556 and GSE101521) were retrieved from the GEO database. Machine learning analysis was used to differentiate three regrouped gene expression profiles, i.e., MDD with suicide, MDD without suicide, and suicide without depression. Weighted correlation network analysis (WGCNA) was further conducted to identify the key modules and hub genes significantly associated with each of these three sub-phenotypes. TissueEnrich approaches were used to find the essential brain tissues and the difference of tissue enriched genes between depression with or without suicide. Dysregulated gene expression cross two variables, including phenotypes and tissues, were determined by global analysis with Vegan. RRHO analysis was applied to examine the difference in global expression pattern between male and female groups. Using the optimized machine learning model, several ncRNAs and mRNAs with higher AUC and MeanDecreaseGini, including GCNT1P1 and AC092745.1, etc., were identified as potential molecular targets to distinguish suicide with, or without MDD and depression without suicide. WGCNA analysis identified some key modules significantly associated with these three phenotypes, and the gene biological functions of the key modules mainly relate to ncRNA and miRNA processing, as well as oxidoreductase and dehydrogenase activity. Hub genes such as RP11-349A22.5, C20orf196, MAPK8IP3 and RP11-697N18.2 were found in these key modules. TissueEnrich analysis showed that nucleus accumbens and subiculum were significantly changed among the 6 brain regions studied. Global analysis with Vegan and RRHO identified PRS26, ARNT and SYN3 as the most significantly differentially expressed genes across phenotype and tissues, and there was little overlap between the male and female groups. In this study, we have identified novel gene targets, as well as annotated functions of co-expression patterns and hub genes that are significantly distinctive between depression with suicide, depression without suicide, and suicide without depression. Moreover, global analysis across three phenotypes and tissues confirmed the evidence of sex difference in mood disorders.
Similar content being viewed by others


Introduction
Major depressive disorder (MDD), a common mental illness, has become the leading cause of disability and disease burden worldwide1. MDD is highly associated with suicide. On the one hand, approximately 15% of MDD patients have their lifetime risk for suicide2; on the other hand, nearly 60% of individuals who died by suicide had a history of mood disorders3. Historical and recent data have shown that over 85% individuals who died by suicide had psychiatric disorders, including affective disorders, particularly in females4,5,6. Some MDD risk factors, including childhood adversity, stress, hopelessness, and aggressivity, may contribute to the activation of suicide ideation across a wide range of the lifespan7,8,9,10. Although some previous studies on the etiology of suicide have suggested that some neurobiological abnormalities of suicide ideation and suicidal behavior are independent of psychiatric disorders, extensive postmortem brain studies have revealed that some changes at the molecular, cellular and circuit levels that may trigger suicide, have both independent and additive effect on depression through different pathways and brain circuitries11,12,13,14.
To date, large-scale genome wide association studies (GWAS) of MDD have produced some consistent genetic risk factors that contribute to susceptibility to depression15,16,17,18,19. For example, a meta-analysis of 3 largest MDD GWASs has identified 102 independent variants, 269 genes, and 15 gene sets, which have shown genome-wide significant association with depression20. The most recent meta-analysis of MDD GWAS has included 366,434 cases and 847,433 controls with both Caucasian and African background, has uncovered 233 independent significant SNPs at 183 genomic risk loci21. Subsequently, the integration of GWAS results and transcriptional data was further conducted in the context of blood and postmortem brain tissues of depression. However less such genome-wide significant results were found in suicide22,23,24,25. From the view of multi-omics on postmortem brain tissues, transcriptomic and epigenetic changes in depression and suicide have been recapitulated using postmortem brain tissue as study materials that directly reflect changes relevant to the neurobiology of suicide and psychiatric disorders26,27,28,29,30. By examining some potential functional candidate genes, abnormalities in the serotonergic, glutamatergic, and GABAergic systems, as well as the hypothalamic–pituitary–adrenal axis for stress response, and the inflammatory pathways, have been detected in depressed or suicidal brain tissue31,32,33,34. More recent transcriptomic studies on different brain regions of depressed and suicidal individuals have not only confirmed the previous findings on GABA- and glutamate-related genes, etc., but also have discovered some novel genes and important signaling pathways35,36,37,38,39,40,41,42. Among these transcriptomic studies, a large-scale transcriptomic study of different brain tissues from depressed patients, who died from suicide, first directly explored the molecular mechanism of sexual dimorphism in MDD. This study has surprisingly shown that very little overlapping of differentially expressed genes was shared between males and females, both in depressed humans and with stressed mice43. Meanwhile, the sex-specific hub genes from the gene co-expression patterns (modules), e.g., DUSP6 and EMX1, were involved to promote stress vulnerability in female and male mice, separately43. Another transcriptome study of depression and suicide examined the genome wide exon gene and microRNA expression profile in the dorsal lateral prefrontal cortex of different types of postmortem brains, including non-psychiatric controls, MDD with suicide, and MDD non-suicides. The results of mRNA-seq GO analysis demonstrated that the genes involved microglial, endothelial, and glial cell DNA-dependent ATPase activity contributed to MDD and suicide44. Considering that a significant fraction of postmortem human brain donors with depression died of suicide, here we have performed a series of bioinformatics analyses with these two high-quality datasets to identify the novel targets of suicide, which are distinctive from the main pathways for MDD.
Subjects, data and methods
Postmortem mRNA expression data and reclassification of sub-phenotypic groups
Two previously reported postmortem mRNA expression profiling datasets for MDD studies were retrieved from the GEO database (GSE102556 and GSE101521). Besides of the corresponding gene expression data, we also obtained demographic data, as well as suicide information from each subject. We summarized the demographics and clinical characteristics, and re-grouped the subjects, based information from concomitant MDD and suicide information from these two datasets (Supplementary Table 1).
The GSE102556 study (PMID: 28825715) included 48 human subjects (age: 47 ± 15 years, F:M = 22:26); each subjects had 6 postmortem brain regions, including Anterior insula (Ant), orbitofrontal (BA11), cingulate gyrus 25 (BA25), dorsolateral prefrontal cortex (BA8/9), nucleus accumbens (Nac) and subiculum (Subic). Based on the occurrence of suicide and MDD diagnosis, the subjects from the GSE102556 study, i.e., the Suicide Group, were reclassified into three groups: suicide with MDD (S + D_1, n = 26, age: 45 ± 14 years, F:M = 13:13), suicide without MDD (S-D_1, n = 11, age: 48 ± 17 years, F:M = 5:6), and non-suicide without MDD (CTR_1, n = 11, age: 57 ± 16 years, F:M = 4:7).
The GSE101521 study (PMID: 27528462), i.e., the Depression Group, included 59 subjects (age 49 ± 21 years, F:M = 17:42) with one postmortem brain region of dorsolateral prefrontal cortex (BA9), which were reclassified as MDD with suicide (D + S_2, n = 21, age: 52 ± 22 years, F:M = 8:13), MDD without suicide (D-S_2, n = 9, age: 58 ± 16 years, F:M = 3:6), and control (CTR_2, n = 29, age: 44 ± 21 years, F:M = 6:23). The clinical and demographic information of these participants in this study was collected by experienced psychiatrists and care-providers; and the clinical diagnostic criteria for the disease were referred to DSM-IV and SCID-1D.
The gene expression data from one overlapped brain region, i.e., the dorsolateral prefrontal cortex (BA9), from these two datasets were further merged and normalized according to the standard protocol; and the batch effect was removed by the limma package (v3.36.5)45. An aligned and normalized gene expression data-frame was generated for each subject. Principal variance component analysis with ExpressionNormalizationWorkflow was performed to estimate the data structural variances, and the significant expression principal components (PCs), adjusted for known covariates, including sex, age, postmortem interval (PMI), RNA integrity number (RIN)46.
Differential gene expression analyses
To find out the characteristic gene expressions as candidate mRNA for different sub-phenotypes, machine learning analyses were performed for the merged gene expression dataset from the BA9 region. First, the normalized TPM (transcripts per million) matrices were proceeded using linear fit in the limma package to perform differential expression analyses of three comparisons, including S + D_vs_CTR, S-D_vs_CTR, and D-S_vs_CTR. Genes with a false discovery rate (FDR) less than 0.05 were defined as differentially expressed genes (DEGs), as previously reported47. The overlapping DEGs between each comparison were plotted as venn diagram.
Machine learning analyses
Then, the genes that were specifically enriched with one sub-phenotype were considered as candidate molecules; and the machine learning model was built with randomforest algorithm as previously reported48. During the training and testing stage, all subjects were divided into two classes using the createDataPartition function, about 80% of the participants were assigned to the training part, while the remaining 20% of the subjects were assigned to the testing part. Meanwhile, we optimized the two important parameters of mtry and ntree of the machine learning model to obtain the smallest estimation error rate so that it could have discriminate between classifiers. Among these two parameters, mtry represent the number of variables randomly sampled when building a decision tree branch in random forest modeling; and the optimal value of mtry parameter reflects as the lowest prediction error rate of the model. While ntree stands for the number of decision trees. In general, the lower ntree value will produce higher error rate while the higher ntree value will increase the complexity of model and reduce the prediction efficiency. Therefore, we first determined the optimal "mtry" parameter and the number of ntrees, based on the above candidate predictors as variables and tuneRF function. Subsequently, tenfold cross-validation was performed to avoid overfitting the model while determining the number of DEGs for the optimal classifier. Here, we introduced the Gini index as an evaluation index for the importance of the features (DEGs) the value of which was proportional to the number of sample splits. Usually, the higher value of the Gini index for a feature (DEGs), the more important it is. The genes having higher Gini Index were selected to input the random forest model and to calculate the area under the curve (AUC) values using the true class labels of half of the tested samples.
Weighted gene co-expression network analysis (WGCNA)
In order to further identify distinctive gene networks and pathways associated with three subphenotypes, we performed a weighted gene co-expression network analysis (WGCNA). First, a standard linear two-way analysis of variance was performed with aov function independently on the data for each gene49,50. The effects examined for each gene were difference among three phenotypic groups. Known covariates (e.g., sex, age, PMI, RIN) were adjusted with glm function for each gene, and significant genes in the two datasets were used for the following WGCNA analysis. WGCNA was conducted to identify key modules and hub genes significantly associated with a particular phenotype. Following the WGCNA tutorials and our previously reported studies, the soft-threshold β was selected using the pickSoftThreshold function based on the uncertain scale-free topology51,52,53. First, the similarity matrix was obtained by calculating the correlation coefficient between genes and then converted into an adjacency matrix with the function pickSoftThreshold, which can determine the best soft threshold/value and average degree of connectivity on the conditions of scale-free network construction. Here, we chose the value equal to 7 as the soft threshold at this point of the fitting index just reaches 0.85. Then, we transformed the adjacency matrix into the topological overlap matrix by means of the function TOMsimilarity. From its dissimilarity of topological overlap matrix, genes with similar expression patterns were divided into the same module labeled with a certain color, where minModuleSize of each module can be a set 30 genes as default number, and the genes in the gray module are not involved in subsequent research. Subsequently, we calculated the Moduleeigengene (ME) of each module which was defined as the first principal component of all gene expression data in the specific module and represented the overall level of gene expression. The spearman correlation coefficient from ME and phenotype trait association analysis was used to analyze the correlation between the module and phenotypic characteristics. Then, we selected the key modules with the largest correlation coefficients and the smallest p values by observing the results in the module feature map. Finally, chooseTopHubInEachModule function was utilized to identify the hub genes in the associated modules, which were visualized in the form of network graphs plotted with igraph package. To annotate the biological functions of key modules, clusterProfile package was used to enrich the genes in the modules into biological pathways and pathways.
Tissue-specific and sex-specific gene expression for suicide
We further explored the GSE102556 dataset based on re-organized dataframe based on suicide with and without MDD sub-phenotypes for tissue-enrichment and sex-specific effects on gene expression. First, we analyzed the transcript ID expression changes across six types of brain tissues using the aov function, which represents the similarity and difference between different tissues. To be specific, it will be spitted into three different dataframes, corresponding to 3 phenotype groups. ANOVA was performed to test for differences among 6 independent tissue groups for each gene. The p value of ANOVA analysis was corrected by cofounding factors. This cross-tissue analysis was individually performed in three phenotypic groups (S+D_1, S−D_1 and CTR_1). Also, the significant level of each gene will be acquired as differentially expressed genes (DEGs), which showed the variance among the six regions. To further explore the enriched tissues of these significant genes, we conducted the tissue-specific gene enrichment analysis using the TissueEnrich package54,55. Referred as the TissueEnrich handbook, the dataframe containing expression information (rows as genes, columns as tissues) was input into SummarizedExperiment function. The teGeneRetrieval function was used to calculate tissue-specific gene enrichment. According to TissueEnrich tutorials, the default threshold can be optional changes to alter the degree of tissue specificity of the gene. In current study, we set the threshold as 4-fold changes, which means genes from tissue enhanced groups which reach at least 4-fold compared to the average levels in all other tissues, are considered brain tissue-specific genes in suicide with depression and suicide without depression groups.
In order to investigate the global difference between the transcriptional signature of whole brain tissues under the two-layers of phenotypes and tissues, Analysis of Similarity (ANOSIM) was performed with the function anoism from the Vegan package to simultaneously detect the dysregulation of gene expression in the multidimensional scaling across three phenotypes and six tissues (cross phenotype and tissue analysis)56. As a non-parametric statistical test, ANOSIM has been widely used in the field of ecology; and the objective of ANOSIM mainly operates on a ranked dissimilarity matrix instead of raw data. First, one dataframe was produced containing the mRNA expression level for the total of samples from three groups and 6 tissues. Then, the function vegdist will produce a suitable dissimilarity matrix from raw dataframe; and anosim function directly operates on dissimilarity matrix with permutation correction. If the entire brains from the three phenotype groups do differ, then the difference between the three groups should be greater than the difference within the group. The heteromorphic statistic R (R = (rB − rW)/(N(N − 1)/4)) is calculated on the basis of difference in mean ranking between groups (rB) and within groups (rW). The statistical significance of the observed R was calculated by permuting the grouping vector to obtain the empirical distribution of R under the null model. We then divided all subjects into male and female groups and performed ANOSIM analysis in each of the male and female groups, as well as calculated the global significance level across phenotypes and tissues in each of the male and female groups.
The extent of overlap of dysregulated genes highlighted in male and female subjects were examined by performing unbiased RRHO (v1.26.0), as described previously57,58,59,60. With a threshold-free method, RRHO analysis mainly estimate overlap between two ranked lists of genes. To perform RRHO analysis, the two gene sets were firstly ranked at the genome-wide scale according to their − log10 (P value) and the direction of change revealed by differential expression analyses. Secondly, a series of hypergeometric p values were calculated for each gene by sliding the rank threshold to examine the significance of overlapping genes above the expected threshold at each ranking site. Lastly, the hypergeometric p values of genes were plotted on a heatmap.
Ethical approval
The Ethics Committee of the Shanghai University of Traditional Chinese Medicines waived the ethics approval and consent for the collection, analysis and publication of the transcriptional data for this study.
Results
Identification of DEGs, co-expression patterns, and hub genes for suicide with depression, suicide without depression and depression without suicide
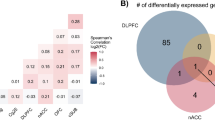
The significant p value of each transcript ID is sequentially calculated with aov function to show the changes of dysregulated expression genes across three phenotypic groups (Suicide Group: CTR_1, S+D_1, and S−D_1; Depression Group: CTR_2, D−S_2, D+S_2). This cross-phenotype analysis was separately conducted in the six brain regions in Suicide Group and one region in Depression Group. Then, the ExpressionNormalizationWorkflow was utilized to determine the variance of the principal components of the DEGs expression data that were impacted by demographic and other confounding factors. The results suggested sex, age, alcohol abuse and RNA integrity had a significant effect on the principal components (p < 0.05). Therefore, these demographic factors were used as confounding factors to correct for any significant difference. After correction with logistic regression analysis, the total number of 2467 DEGs were screened in the Depression Group (Supplementary Table 2; Supplementary Fig. 1). Regarding the number of DEGs in the six tissues in the Suicide Group, the most DEGs (n = 3773) were identified in the dorsolateral prefrontal cortex while the least DEGs (n = 1066) in the subiculum (Supplementary Table 3; Supplementary Fig. 2). Some genes, such as DUSP6 and mitogen-activated protein kinase genes, have been previously reported in the depression and suicide study while more novel genes were found. In general, the changes trend for most genes were observed to be upregulated in the Suicide Group (Supplementary Table 3). The similarities and differences genes of the 6 regions were shown in venn diagram (Fig. 1A). After examining gene expression in the 6 tissues, two transcripts containing RP11-459O16 belonging to the lncRNA family and CH25H coding with cholesterol 25-hydroxylase have significant changes overlapping in the 6 regions, suggesting that these two genes may play a contributory role in the connectivity of different brain tissues.

Similarity and overlapping in the DEGs between different tissues and diseases. (A) DEGs from the cross-phenotype analysis. Different colors represent different tissues, overlapping parts represent genes present in all tissues, parts of not overlap represent DEGs that are specific to the tissues, and numbers indicate the number of DEGs in the corresponding tissue. (B) Venn diagram of the different phenotypes. Colors represent each phenotype, overlapping parts represent shared genes among phenotypes, parts of not overlapping represent DEGs that are specific to the diseases, and numbers indicate DEGs numbers of different phenotypes.
To further investigate the co-expression genes as a module significantly associated with certain phenotype, WGCNA analysis was performed with the differential expression genes in the dorsolateral prefrontal cortex regions from these two groups. As described in the methods, 4 modules in the Suicide Group (S+D_1, S–D_1 and CTR_1) and 9 modules in the Depression Group (D+S_2, D–S_2 and CTR_2) were assigned. Subsequent analysis showed that each module was correlated with different phenotypes. Here, we focused on the modules having significant association with the phenotype of depression with suicide and suicide without depression. For example, the midnightblue module that was classified as the top positive module in the Suicide Group, showed a significant association with suicide without depression (p value = 1E−12) (Fig. 2A). Interestingly, the top significant GO annotated functions mainly relate to the activity of different types of oxidoreductases and dehydrogenase, including testosterone-, aldehyde-, and retino, etc., as well as glucocorticoid receptor binding and Golgi terms (Fig. 2C). On the other hand, the blue module in the Suicide Group, which is the top negatively correlated with suicide without depression, is mainly involved in the ncRNA, miRNA and dsRNA processing activity (Fig. 2A,D). For the Depression Group, the pink module of biological functions which focuses on the biological functions of postsynaptic synapse, has a significant negative association with depression without suicide (Fig. 2B,E). In contrast, orange module, as positive module for depression without suicide, mainly mediates into the signaling pathway for primary cilia and retinal sensitivity (Fig. 2B,F). Key hub genes were calculated based on the connectivity level of the co-expression network of each module (Supplementary Table 4). RP11-349A22.5 in the blue module and C20orf196 in the midnightblue module were identified as hub gene having high connectivity with nodes in a module and these two hub genes have been shown to have a key function in differentiating suicide without depression while MAPK8IP3 in the pink module and RP11-697N18.2 in the orange module were found to be hub genes in the module significantly associated with depression without suicide (Supplementary Fig. 3).

Module-trait relationship analysis with WGCNA and the KEGG pathway analysis for the correlated modules. (A) Module-phenotype relationship for the assigned module and different phenotypes from Suicide Group. Values in the figure indicate the correlation coefficient between module and phenotypes. The midnightblue module was positively correlated with suicide without MDD (r = 0.82, p value = 1E−12) while the blue module was negatively correlated with suicide without MDD (r = − 0.44, p value = 0.022). (B) Module-phenotype relationship in the Depression Group. The two modules including the orange and pink module were the most significantly associated with MDD without suicide (orange module: r = − 0.62, p value = 2E−7; pink module: r = 0.54, p value = 1E−5). (C) The enriched GO functions of midnightblue module from Suicide Group. (D) The overrepresented GO functions of the blue module from Suicide Group. (E) Gene ontology analysis of the pink module from Depression Group. (F) Gene ontology analysis of the orange module from Depression Group.
Screening of important molecules as candidate targets for three sub-phenotypes
To maximally identify the potential targets for depression with suicide, we merged the subjects with depression with suicide shared these two datasets to enlarge the sample size. Also, the further PCA analysis found no stratification of cohort structure (Supplementary Fig. 4). Three comparisons, including suicide with depression and control, suicide without depression and control, depression without suicide and control, were used to conduct the comparison and contrast analysis (Fig. 1B). The total number of 3373, 1112 and 2991 DEGs were specifically assigned in the not overlapping section with suicide with depression, suicide without depression and depression without suicide, respectively (Fig. 1B; Supplementary Table 5). Then, the expression level of these specific DEGs was used to establish the machine learning model while the subjects were classified into two classes (disease state and healthy control). As described in the methods, the parameters were optimized to ensure that the model had the lowest prediction error rate. Finally, the MeanDecreaseGini of GCNT1P1 and AC092745.12 ranked as the top important candidate genes for suicide with depression (AUC = 0.90). Meanwhile, lncRNA AC010084.1 and RP11-1C8.7 had the highest Gini index in the S–D group, suggesting the possible link of suicide without MDD with these lncRNAs (AUC = 0.86). For depression without suicide, the MPOL1 and SLC2A13 gene had the highest Gini index score and may be the candidate molecules (Table 1; Supplementary Fig. 5).
Identification of tissue-specific genes in suicide group
To examine some genes enriched in specific brain regions, cross-tissue analysis was performed calculate the changes level of all transcripts in the group of control, suicide with depression, and suicide without depression, respectively (Supplementary Table 6; Supplementary Fig. 6). Based on these dysregulated genes from the cross-tissue analysis, the brain tissue specific genes were screened out by means of TissueEnrich package. The results of TissueEnrich demonstrated that in the two brain regions, including Nac and subic, 14 and 16 genes, respectively, were found to reach tissue- enhanced level, that is, these two brain areas play a critical role in the development of depression (Table 2; Supplementary Table 7). The gene MYO3A encoding myosin IIIA, was highly enriched in the Nac tissues of all three groups, which suggested that MYO3A gene as Nac tissue specific gene is unrelated to the phenotype. At the same time, the expression of TNFRSF8 gene was enhanced into the Nac of the control group and the group of suicide with MDD, but not in the group of suicide without MDD. Another gene, TARID transcript as a non-coding RNA, was found in the Nac of control and suicide without MDD, but not in the suicide with MDD. We speculated that TNFRSF8 and TARID were considered as not only the tissue specific genes but also phenotype-specific genes. For the subic tissue, no genes as tissue-enhanced genes were shared among three groups. In addition, CLIC6, encoding chloride intracellular channel 6, enriched in the subic of both suicide without MDD and control, but was absent in the suicide with MDD.
Sex specific signatures of expression patterns in suicides
By means of cross phenotype tissue analysis, we calculated the R and p value of all transcript genes for the total subject group, male group, and female group. We also ranked the statistical R value of all significant transcripts and listed the top genes in Table 3. The top R genes in the mixed gender group (PRS26 (R = 0.37), ARNT (R = 0.32) and SYN3 (R = 0.32), the male group (FBXO7 (R = 0.41), RNU6-951P (R = 0.40) and ACTR8 (R = 0.39)) and the female group (ADI1 (R = 0.64), CH25H (R = 0.63) and CCR1 (R = 0.57), were identified as the most differentially expressed genes across two variables of tissues and phenotypes (Table 3; Supplementary Fig. 7). To identify differences between male and female in global expression patterns across three phenotypes and multiple tissues, the results of rank–rank hypergeometric overlay analysis showed that there was no significant overlap of global gene expression patterns were detected between male and female groups (p value = 0.92) (Supplementary Fig. 8).
Discussion
In this study, we identified some novel targets that can be used to distinguish different types of postmortem brain tissues from depression with or without suicide and suicide without depression. In addition, we also examined the tissue-specific and sex-specific transcriptional patterns between suicide dependent or independent of depression. Here, we selected two classical genome wide expression datasets of postmortem brain tissues from depression or suicide and extracted the demographic information of human postmortem brain tissue about having suicide or non-suicide, from the perspective of commodity phenotypes or single phenotype to find the possible key genes or co-expression patterns. Considering differentially expressed genes may correlate with different phenotypes, brain tissues and sex, various statistical strategies including ANOVA, limma and Vegan were adapted to conduct WGCNA, machine learning and RROH. We found that most of the DEGs, especially the top significant genes, were consistent with previous studies, such as MRPS6, SERPINH1and IL8 gene in GSE101521, DUSP6, EMX1 and MPAK15 gene in GSE102556. In addition, we also identified novel possible transcripts and ncRNA that contribute to the discrimination of depression or suicide.
Postmortem brain tissue as an important material reflects the molecular and cellular changes in brain tissues from suicided victims and controls, which are also features of mental illness11. However, the prominent consideration of postmortem brain studies is the impact of comorbid suicide diagnoses including some common psychiatric disorders and other risk factors that influence the transitions from suicide ideation to suicide attempts61. From the perspective of suicide with psychiatric disorders, Underwood et al.62 investigated brain serotonin transporter, 5-HT1A and 5-HT2A receptor binding in the prefrontal cortex and anterior cingulate cortex of suicide and controls. They reported that lower serotonin transporter levels in suicide were dependent on MDD, and found that higher 5-HT1A binding in suicides was independent of MDD, whereas 5-HT2A binding increased in suicides with MDD or alcoholism. This study suggested serotonin receptor binding in postmortem brain tissue can be a risk factor to separate the effects of suicide from comorbid MDD, AUD, and early life adversity62. In the present study, we explored the suicide- or depression-specific mRNA and mRNA co-expression pattern changes that contribute to development of suicide and depression. From the WGCNA results, the biological functions of co-expression pattern associated with suicide without depression are mainly related to ncRNA processing activity, whereas depression without suicide can be attributed to neuronal synapses, suggesting that epigenetic modification and synaptic dysfunction in brain tissues play an essential role in the development of suicide or depression.
For machine learning analysis, we integrated contrast analyses of multiple comparisons and machine learning to identify candidate targets for a given sub-phenotype. In this process, we first maximally optimized the various parameters for building the training model by means of randomforest and obtained relatively accurate distinguishing capacity. To confirm the distinguishing accuracy of genes with high MeanDecreaseGini, we compared the AUC score between randomforest models of genes with high MeanDecreaseGini and the group of all genes, the results of which shown AUC score of genes with high MeanDecreaseGini were largely enhanced in the S + D_1 (AUC high MeanDecreaseGini genes = 0.90; AUC total genes = 0.81). Intriguingly, we also found majority of these genes with high MeanDecreaseGini belong to the family of non-coding RNAs. Moreover, several ncRNAs including GCNT1P1, AC092745.1, ASH1L-IT1 and RNU6-581P from the S + D_1 group are specifically enriched in the cerebellum and pituitary by the analysis of gene expression levels across different human tissues in the GTEx dataset, suggesting that ncRNAs that are highly expressed in the cerebellum and pituitary may be used as specific targets for suicide and MDD63. Currently, machine learning analysis are used for diagnosis, prognosis, treatment decisions, and detection of biomarker for suicide64,65. To predict suicide attempts and suicide deaths, the prediction model was developed using the penalized LASSO method with health record data and self-report questionnaires from about 3 million patients, and the results have shown an accuracy ranging between 0.83 and 0.8566,67. Meanwhile, machine learning was used for exploring biomarkers of the antidepressant by analyzing the expression of microRNA in previous MDD studies68.
TissueEnrich was used to find out the essential tissues for the development of depression and identify tissue-specific genes, and co-expression patterns associated with tissues. Interestingly, the enhanced genes in the TissueEnrich suggested that the Nac region may be the most important tissue contributing to the pathophysiology and symptomatology of depression and suicide. Under normal conditions, Nac function is related to the brain’s limbic and reward circuits and the uptake of glutamatergic and dopaminergic signals from other brain tissues69. Evidence from the rodent depression model and the antidepressant-like effects also support that Nac tissues are involved in the impaired motivation, anhedonia, and decreased energy levels which are core symptoms of depression70. Despite that genome-wide transcriptional study supports that several molecular changes in the Nac of depressive like behavior animal models, we further validated the importance of Nac and subic for suicide or depression71.
We have uncovered the global landscape of distinct expression patterns for different tissues and individuals in depression and suicide. In recent years, the development of multi-dimension informatics approaches including vegan and Tensorflow, has enabled the global investigation of significantly dysregulated gene expression across multiple variables, and have identified key genes for the concomitant diseases. The subsequent RRHO analysis58 further confirmed that the global gene expression patterns have little overlap between the male and female group under the situations of suicide independent depression or suicide comorbid depression, which is consistent with the conclusions of previous transcriptomic studies in depression43,59,72,73.
At present, transcriptional studies from candidate genes to genome-wide approaches have also identified some dysregulated genes associated with sex difference in MDD. Some candidate genes, from serotonergic system to GABA synthesizing enzymes, were found to have altered expression associated with sex difference in MDD 72. Particularly some key genes identified from transcriptomic studies, e.g., DUSP6, EMX1, LINC00473, as potential important drivers for sex difference, directly involved in the brain molecular changes in depressed men and women43. Additionally, novel approaches including global analysis in present study and gene co-expression network analysis and deconvolution analyses provided more convincing evidence that the existence of sex differences across the brain in the MDD. About the underlying mechanisms on sex difference at the behavioral or transcriptional levels, evidence has suggested that the interplay between sex chromosomes, development gonadal hormones and adult hormone exposure could influence social behavior, alcohol abuse, habit formation, aggressive and parenting behavior, and gene expression, which could mediate the different mood changes in male and female MDD patients72,74.
In summary, we investigated different transcriptomic datasets to uncover differential gene targets and pathways among subgroups of depression with suicide, suicide without depression and depression without suicide. We also investigated the co-expression patterns and hub genes significantly associated with these three sub-phenotypes. Meanwhile, global analysis across three phenotypes and tissues confirmed evidence for sex difference in mood disorders. Our study is still limited by the sample size in each subgroup, as well as limited detailed clinical information and medical history on postmortem brain tissues.
URLs
-
1.
Heatmap: https://cran.r-project.org/web/packages/gplots/index.html.
-
2.
WGCNA analysis: https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/Tutorials/.
-
3.
Machine learning analysis Rando: https://topepo.github.io/caret/model-training-and-tuning.html, https://rpubs.com/phamdinhkhanh/389752.
-
4.
TissueEnrich analysis: https://bioconductor.org/packages/release/bioc/vignettes/TissueEnrich/inst/doc/TissueEnrich.html.
-
5.
Vegan analysis: https://cran.r-project.org/web/packages/vegan/vegan.pdf.
-
6.
RRHO analysis: https://systems.crump.ucla.edu/rankrank/rankranksimple.php.
Data availability
The authors declare that all the data supporting the findings of this study are available from the GEO database (GSE102556 and GSE101521); and some secondary results can be found in its supplementary information files.
Code availability
The authors declare that all the codes supporting the findings of this study are available from the corresponding author on reasonable request.
References
WHO. Depression (World Health Organization, 2020).
Orsolini, L. et al. Understanding the complex of suicide in depression: From research to clinics. Psychiatry Investig. 17, 207–221. https://doi.org/10.30773/pi.2019.0171 (2020).
Mann, J. J. The neurobiology of suicide. Nat. Med. 4, 25–30. https://doi.org/10.1038/nm0198-025 (1998).
Arsenault-Lapierre, G., Kim, C. & Turecki, G. Psychiatric diagnoses in 3275 suicides: A meta-analysis. BMC Psychiatry 4, 37. https://doi.org/10.1186/1471-244x-4-37 (2004).
Seiffge-Krenke, I. Depression in children and adolescents: Prevalence, diagnosis, etiology, gender differences and therapeutic approaches. Prax Kinderpsychol. Kinderpsychiatr. 56, 185–205. https://doi.org/10.13109/prkk.2007.56.3.185 (2007).
Mazza, M. G. et al. Anxiety and depression in COVID-19 survivors: Role of inflammatory and clinical predictors. Brain Behav. Immunol. 89, 594–600. https://doi.org/10.1016/j.bbi.2020.07.037 (2020).
Turecki, G. et al. Suicide and suicide risk. Nat. Rev. Dis. Primers 5, 74. https://doi.org/10.1038/s41572-019-0121-0 (2019).
Nock, M. K. et al. Cross-national analysis of the associations among mental disorders and suicidal behavior: Findings from the WHO World Mental Health Surveys. PLoS Med. 6, e1000123. https://doi.org/10.1371/journal.pmed.1000123 (2009).
Charles, J. & Fazeli, M. Depression in children. Aust. Fam. Physician 46, 901–907 (2017).
Zhao, F., Yue, Y., Jiang, H. & Yuan, Y. Shared genetic risk factors for depression and stroke. Prog. Neuropsychopharmacol. Biol. Psychiatry 93, 55–70. https://doi.org/10.1016/j.pnpbp.2019.03.003 (2019).
Pandey, G. N. & Dwivedi, Y. What can post-mortem studies tell us about the pathoetiology of suicide?. Future Neurol. 5, 701–720. https://doi.org/10.2217/fnl.10.49 (2010).
Sher, L. Brain-derived neurotrophic factor and suicidal behavior. QJM 104, 455–458. https://doi.org/10.1093/qjmed/hcq207 (2011).
van Heeringen, K. & Mann, J. J. The neurobiology of suicide. Lancet Psychiatry 1, 63–72. https://doi.org/10.1016/s2215-0366(14)70220-2 (2014).
Almeida, D. & Turecki, G. A slice of the suicidal brain: What have postmortem molecular studies taught us?. Curr. Psychiatry Rep. 18, 98. https://doi.org/10.1007/s11920-016-0736-8 (2016).
Howard, D. M. et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 22, 343–352. https://doi.org/10.1038/s41593-018-0326-7 (2019).
Howard, D. M. et al. Genome-wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nat. Commun. 9, 1470. https://doi.org/10.1038/s41467-018-03819-3 (2018).
Yao, X. et al. Integrative analysis of genome-wide association studies identifies novel loci associated with neuropsychiatric disorders. Transl. Psychiatry 11, 69. https://doi.org/10.1038/s41398-020-01195-5 (2021).
Consortium C. Sparse whole-genome sequencing identifies two loci for major depressive disorder. Nature 523, 588–591. https://doi.org/10.1038/nature14659 (2015).
Wray, N. R. et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 50, 668–681. https://doi.org/10.1038/s41588-018-0090-3 (2018).
Wu, W., Howard, D., Sibille, E. & French, L. Differential and spatial expression meta-analysis of genes identified in genome-wide association studies of depression. Transl. Psychiatry 11, 8. https://doi.org/10.1038/s41398-020-01127-3 (2021).
Levey, D. F. et al. Bi-ancestral depression GWAS in the Million Veteran Program and meta-analysis in > 1.2 million individuals highlight new therapeutic directions. Nat. Neurosci. 24, 954–963. https://doi.org/10.1038/s41593-021-00860-2 (2021).
Li, Q. S. et al. Genome-wide association study meta-analysis of suicide death and suicidal behavior. Mol. Psychiatry https://doi.org/10.1038/s41380-022-01828-9 (2022).
Sokolowski, M., Wasserman, J. & Wasserman, D. Polygenic associations of neurodevelopmental genes in suicide attempt. Mol. Psychiatry 21, 1381–1390. https://doi.org/10.1038/mp.2015.187 (2016).
Flint, J. & Kendler, K. S. The genetics of major depression. Neuron 81, 484–503. https://doi.org/10.1016/j.neuron.2014.01.027 (2014).
Mullins, N. et al. GWAS of suicide attempt in psychiatric disorders and association with major depression polygenic risk scores. Am. J. Psychiatry 176, 651–660. https://doi.org/10.1176/appi.ajp.2019.18080957 (2019).
Zhang, J., Kaye, A. P., Wang, J. & Girgenti, M. J. Transcriptomics of the depressed and PTSD brain. Neurobiol. Stress 15, 100408. https://doi.org/10.1016/j.ynstr.2021.100408 (2021).
Zhou, Y. et al. Suicide and suicide behaviors: A review of transcriptomics and multiomics studies in psychiatric disorders. J. Neurosci. Res. 98, 601–615. https://doi.org/10.1002/jnr.24367 (2020).
Furczyk, K., Schutová, B., Michel, T. M., Thome, J. & Büttner, A. The neurobiology of suicide—a review of post-mortem studies. J. Mol. Psychiatry 1, 2. https://doi.org/10.1186/2049-9256-1-2 (2013).
Barnes, J., Mondelli, V. & Pariante, C. M. Genetic contributions of inflammation to depression. Neuropsychopharmacology 42, 81–98. https://doi.org/10.1038/npp.2016.169 (2017).
Allen, L. & Dwivedi, Y. MicroRNA mediators of early life stress vulnerability to depression and suicidal behavior. Mol. Psychiatry 25, 308–320. https://doi.org/10.1038/s41380-019-0597-8 (2020).
Tripp, A. et al. Brain-derived neurotrophic factor signaling and subgenual anterior cingulate cortex dysfunction in major depressive disorder. Am. J. Psychiatry 169, 1194–1202. https://doi.org/10.1176/appi.ajp.2012.12020248 (2012).
Guilloux, J. P. et al. Molecular evidence for BDNF- and GABA-related dysfunctions in the amygdala of female subjects with major depression. Mol. Psychiatry 17, 1130–1142. https://doi.org/10.1038/mp.2011.113 (2012).
Gray, A. L., Hyde, T. M., Deep-Soboslay, A., Kleinman, J. E. & Sodhi, M. S. Sex differences in glutamate receptor gene expression in major depression and suicide. Mol. Psychiatry 20, 1139. https://doi.org/10.1038/mp.2015.114 (2015).
Goswami, D. B., May, W. L., Stockmeier, C. A. & Austin, M. C. Transcriptional expression of serotonergic regulators in laser-captured microdissected dorsal raphe neurons of subjects with major depressive disorder: Sex-specific differences. J. Neurochem. 112, 397–409. https://doi.org/10.1111/j.1471-4159.2009.06462.x (2010).
Klempan, T. A. et al. Altered expression of genes involved in ATP biosynthesis and GABAergic neurotransmission in the ventral prefrontal cortex of suicides with and without major depression. Mol. Psychiatry 14, 175–189. https://doi.org/10.1038/sj.mp.4002110 (2009).
Sequeira, A. et al. Global brain gene expression analysis links glutamatergic and GABAergic alterations to suicide and major depression. PLoS One 4, e6585. https://doi.org/10.1371/journal.pone.0006585 (2009).
Lalovic, A., Klempan, T., Sequeira, A., Luheshi, G. & Turecki, G. Altered expression of lipid metabolism and immune response genes in the frontal cortex of suicide completers. J. Affect. Disord. 120, 24–31. https://doi.org/10.1016/j.jad.2009.04.007 (2010).
Bernard, R. et al. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Mol. Psychiatry 16, 634–646. https://doi.org/10.1038/mp.2010.44 (2011).
Sequeira, A. et al. Gene expression changes in the prefrontal cortex, anterior cingulate cortex and nucleus accumbens of mood disorders subjects that committed suicide. PLoS One 7, e35367. https://doi.org/10.1371/journal.pone.0035367 (2012).
Venuto, S. et al. TRIM8-driven transcriptomic profile of neural stem cells identified glioma-related nodal genes and pathways. Biochim. Biophys. Acta Gen. Subj. 491–501, 2019. https://doi.org/10.1016/j.bbagen.2018.12.001 (1863).
Chang, L. C. et al. A conserved BDNF, glutamate- and GABA-enriched gene module related to human depression identified by coexpression meta-analysis and DNA variant genome-wide association studies. PLoS One 9, e90980. https://doi.org/10.1371/journal.pone.0090980 (2014).
Duric, V. et al. Altered expression of synapse and glutamate related genes in post-mortem hippocampus of depressed subjects. Int. J. Neuropsychopharmacol. 16, 69–82. https://doi.org/10.1017/s1461145712000016 (2013).
Labonté, B. et al. Sex-specific transcriptional signatures in human depression. Nat. Med. 23, 1102–1111. https://doi.org/10.1038/nm.4386 (2017).
Pantazatos, S. P. et al. Whole-transcriptome brain expression and exon-usage profiling in major depression and suicide: Evidence for altered glial, endothelial and ATPase activity. Mol. Psychiatry 22, 760–773. https://doi.org/10.1038/mp.2016.130 (2017).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. https://doi.org/10.1093/nar/gkv007 (2015).
Karthikeyan, M. Expression Normalization Workflow: Gene Expression Normalization Workflow, R Package Version 1.18.0 (R Foundation For Statistical Computing, 2021).
Roy, B., Wang, Q. & Dwivedi, Y. Long noncoding RNA-associated transcriptomic changes in resiliency or susceptibility to depression and response to antidepressant treatment. Int. J. Neuropsychopharmacol. 21, 461–472. https://doi.org/10.1093/ijnp/pyy010 (2018).
Liao, Q. et al. An integrated method for optimized identification of effective natural inhibitors against SARS-CoV-2 3CLpro. Sci. Rep. 11, 22796. https://doi.org/10.1038/s41598-021-02266-3 (2021).
Zar, J. Biostatistical Analysis (Fourth Edition) 32–47 (Prentice Hall Upper Saddle River, 1999).
Mathew, T. (Taylor & Francis, 2001).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Langfelder, P. & Horvath, S. Fast R Functions for Robust Correlations and Hierarchical Clustering. J. Stat. Softw. 46, 25 (2012).
Wang, Q., Roy, B. & Dwivedi, Y. Co-expression network modeling identifies key long non-coding RNA and mRNA modules in altering molecular phenotype to develop stress-induced depression in rats. Transl. Psychiatry 9, 1–13 (2019).
Jain, A. & Tuteja, G. TissueEnrich: Tissue-specific gene enrichment analysis. Bioinformatics 35, 1966–1967. https://doi.org/10.1093/bioinformatics/bty890 (2018).
Jain, A. & Tuteja, G. TissueEnrich: Tissue-specific gene enrichment analysis. Bioinformatics 35, 1966–1967. https://doi.org/10.1093/bioinformatics/bty890 (2019).
Oksanen, J. et al. (2020).
Bagot, R. C. et al. Circuit-wide transcriptional profiling reveals brain region-specific gene networks regulating depression susceptibility. Neuron 90, 969–983. https://doi.org/10.1016/j.neuron.2016.04.015 (2016).
Plaisier, S. B., Taschereau, R., Wong, J. A. & Graeber, T. G. Rank-rank hypergeometric overlap: Identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res. 38, e169. https://doi.org/10.1093/nar/gkq636 (2010).
Seney, M. L. et al. Opposite molecular signatures of depression in men and women. Biol. Psychiatry 84, 18–27. https://doi.org/10.1016/j.biopsych.2018.01.017 (2018).
Cahill, K. M., Huo, Z., Tseng, G. C., Logan, R. W. & Seney, M. L. Improved identification of concordant and discordant gene expression signatures using an updated rank-rank hypergeometric overlap approach. Sci. Rep. 8, 9588. https://doi.org/10.1038/s41598-018-27903-2 (2018).
McCullumsmith, R. E., Hammond, J. H., Shan, D. & Meador-Woodruff, J. H. Postmortem brain: An underutilized substrate for studying severe mental illness. Neuropsychopharmacology 39, 65–87. https://doi.org/10.1038/npp.2013.239 (2014).
Underwood, M. D. et al. Serotonin receptors and suicide, major depression, alcohol use disorder and reported early life adversity. Transl. Psychiatry 8, 279. https://doi.org/10.1038/s41398-018-0309-1 (2018).
Consortium, G. T. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 348, 648–660. https://doi.org/10.1126/science.1262110 (2015).
Gopalkumar Rakesh, M. D. Suicide prediction with machine learning. Am. J. Psychiatry Residents J. 12, 15–17. https://doi.org/10.1176/appi.ajp-rj.2017.120105 (2017).
Rajpurkar, P. et al. Evaluation of a machine learning model based on pretreatment symptoms and electroencephalographic features to predict outcomes of antidepressant treatment in adults with depression: A prespecified secondary analysis of a randomized clinical trial. JAMA Netw. Open 3, e206653–e206653. https://doi.org/10.1001/jamanetworkopen.2020.6653 (2020).
Simon, G. E. et al. Predicting suicide attempts and suicide deaths following outpatient visits using electronic health records. Am. J. Psychiatry 175, 951–960. https://doi.org/10.1176/appi.ajp.2018.17101167 (2018).
Mullah, M. A. S., Hanley, J. A. & Benedetti, A. LASSO type penalized spline regression for binary data. BMC Med. Res. Methodol. 21, 83. https://doi.org/10.1186/s12874-021-01234-9 (2021).
Qi, B., Fiori, L. M., Turecki, G. & Trakadis, Y. J. Machine learning analysis of blood microRNA data in major depression: A case–control study for biomarker discovery. Int. J. Neuropsychopharmacol. 23, 505–510. https://doi.org/10.1093/ijnp/pyaa029 (2020).
Salgado, S. & Kaplitt, M. G. The nucleus accumbens: A comprehensive review. Stereotact. Funct. Neurosurg. 93, 75–93. https://doi.org/10.1159/000368279 (2015).
Nestler, E. J. Role of the brain’s reward circuitry in depression: Transcriptional mechanisms. Int. Rev. Neurobiol. 124, 151–170. https://doi.org/10.1016/bs.irn.2015.07.003 (2015).
Krishnan, V. & Nestler, E. J. Animal models of depression: Molecular perspectives. Curr. Top. Behav. Neurosci. 7, 121–147. https://doi.org/10.1007/7854_2010_108 (2011).
Seney, M. L., Glausier, J. & Sibille, E. Large-scale transcriptomics studies provide insight into sex differences in depression. Biol. Psychiatry 91, 14–24. https://doi.org/10.1016/j.biopsych.2020.12.025 (2022).
Rainville, J. R., Lipuma, T. & Hodes, G. E. Translating the transcriptome: Sex differences in the mechanisms of depression and stress. Revisited. Biol. Psychiatry 91, 25–35. https://doi.org/10.1016/j.biopsych.2021.02.003 (2022).
Lima, S., Sousa, N., Patrício, P. & Pinto, L. The underestimated sex: A review on female animal models of depression. Neurosci. Biobehav. Rev. 133, 104498. https://doi.org/10.1016/j.neubiorev.2021.12.021 (2022).
Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (31871281), Shanghai Municipal Health Commission (2020XGKY12) and Scientific Research Foundation for Advanced Talents of Shanghai University of Traditional Chinese Medicine.
Author information
Authors and Affiliations
Contributions
Q.W., L.X. designed the project, checked and edited the manuscript. Material preparation and data collection and analysis were performed by S.P. and Y.Z. All authors have read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Peng, S., Zhou, Y., Xiong, L. et al. Identification of novel targets and pathways to distinguish suicide dependent or independent on depression diagnosis. Sci Rep 13, 2488 (2023). https://doi.org/10.1038/s41598-023-29101-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-29101-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
