
Abstract
The human SBF1 (SET binding factor 1) gene, alternatively known as MTMR5, is predominantly expressed in the brain, and its epigenetic dysregulation is linked to late-onset neurocognitive disorders (NCDs), such as Alzheimer’s disease. This gene contains a (GCC)-repeat at the interval between + 1 and + 60 of the transcription start site (SBF1-202 ENST00000380817.8). We sequenced the SBF1 (GCC)-repeat in a sample of 542 Iranian individuals, consisting of late-onset NCDs (N = 260) and controls (N = 282). While multiple alleles were detected at this locus, the 8 and 9 repeats were predominantly abundant, forming > 95% of the allele pool across the two groups. Among a number of anomalies, the allele distribution was significantly different in the NCD group versus controls (Fisher’s exact p = 0.006), primarily as a result of enrichment of the 8-repeat in the former. The genotype distribution departed from the Hardy–Weinberg principle in both groups (p < 0.001), and was significantly different between the two groups (Fisher’s exact p = 0.001). We detected significantly low frequency of the 8/9 genotype in both groups, higher frequency of this genotype in the NCD group, and reverse order of 8/8 versus 9/9 genotypes in the NCD group versus controls. Biased heterozygous/heterozygous ratios were also detected for the 6/8 versus 6/9 genotypes (in favor of 6/8) across the human samples studied (Fisher’s exact p = 0.0001). Bioinformatics studies revealed that the number of (GCC)-repeats may change the RNA secondary structure and interaction sites at least across human exon 1. This STR was specifically expanded beyond 2-repeats in primates. In conclusion, we report indication of a novel biological phenomenon, in which there is selection against certain heterozygous genotypes at a STR locus in human. We also report different allele and genotype distribution at this STR locus in late-onset NCD versus controls. In view of the location of this STR in the 5′ untranslated region, RNA/RNA or RNA/DNA heterodimer formation of the involved genotypes and alternative RNA processing and/or translation should be considered.
Similar content being viewed by others

Introduction
While of vast evolutionary and biological implications1,2,3,4,5,6,7,8, short tandem repeats (STRs) remain an underappreciated topic in comparison to single nucleotide substitutions9,10, partly because of their repetitive nature and hardship of accurate allele calling with the currently available methods.
Among various categories of STRs, CGG/GCC repeats are overrepresented in the exons of the human genome, and are mainly focused on because of their involvement in neurological disorders11,12,13,14. The human gene, SBF1 (SET binding factor 1), also known as MTMR5 (Myotubularin-related protein 5) contains an annotated (GCC)-repeat of 9-repeats in the 5′ untranslated region (UTR), between + 1 to + 60 of the transcription start site (TSS) (SBF1-202 ENST00000380817.8), which is in the top 1 percentile of (GCC)-repeats with respect to length15. SBF1 is located at the extreme end of the long arm of chromosome 22 (22q13.33), and across all human tissues, reaches maximum expression in the cerebral cortex (https://www.proteinatlas.org/ENSG00000100241-SBF1/tissue). In comparison with other primate species, SBF1 reaches maximum expression quantiles in the human brain and skeletal muscle (https://www.ncbi.nlm.nih.gov/IEB/Research/Acembly)16. In line with the above, aberrant regulation of the gene networks in which SBF1 plays a role has been reported in late-onset neurocognitive disorders (NCDs), such as Alzheimer’s disease (AD)17.
Here we sequenced the SBF1 (GCC)-repeat in a sample of humans, consisting of late-onset NCDs and controls, and performed structural and accessibility analysis of exon 1 (encompassing this repeat) with various (GCC) repeats. We also studied the status of this (GCC)-repeat across vertebrates.
Materials and methods
Subjects
Five hundred forty-two unrelated Iranian subjects of ≥ 60 years of age, consisting of late-onset NCD patients (DSM-5) (N = 260) and controls (N = 282) were recruited from the provinces of Tehran, Qazvin, and Rasht. In each NCD case, the Persian version of the Abbreviated Mental Test Score (AMTS)18,19 was implemented (AMTS < 7 was an inclusion criterion for NCD), medical records were reviewed in all participants, and CT-scans were taken where possible. Furthermore, in a number of subjects, the Mini-Mental State Exam (MMSE) Test20 was implemented in addition to the AMTS. A score of < 24 was an inclusion criterion for NCD. The Persian version of the AMTS is a valid cognitive assessment tool for older Iranian adults, and can be used for NCD screening in Iran18. The onset of neurocognitive impairment was also investigated by clinical interviews, which confirmed the occurrence of those symptoms at ≥ 60 years. The control group was selected based on cognitive AMTS of > 7 and MMSE > 24, lack of major medical history, and normal CT-scan where possible. The cases and controls were matched based on age, gender, and residential district. The subjects' informed consent was obtained (from their guardians where necessary) and their identities remained confidential throughout the study. The research was approved by the Ethics Committee of the Social Welfare and Rehabilitation Sciences, Tehran, Iran, and was consistent with the principles outlined in an internationally recognized standard for the ethical conduct of human research. All methods were performed in accordance with the relevant guidelines and regulations.
Allele and genotype analysis of the SBF1 (GCC)-repeat
Genomic DNA was obtained from peripheral blood using a standard salting out method. PCR reactions for the amplification of the SBF1 (GGC)-repeat were set up with the following primers:
-
Forward: TCTGGACCAATGGAGATGCG
-
Reverse: GAAGTAGTCCGCGAGCCG
PCR reactions were carried out in a final volume of 20 µl, at a final concentration of 30% high-GC buffer, in a thermocycler (Peqlab-PEQStar) under the following conditions: initial denaturation at 95 °C for 5 min, 40 cycles of denaturation at 95 °C for 45 s, annealing at 55 °C for 45 s, and extension at 72 °C for 1 min, and a final extension at 72 °C for 10 min. All samples included in this study were sequenced by the forward primer, using an ABI 3130 DNA sequencer (Suppl. 1).
Statistical analysis
The SPSS Fisher’s exact test was used to compare allele and genotype distribution between NCD and control groups. Fisher’s exact test was also used for the 6/8 versus 6/9 genotypes. The Hardy–Weinberg principle (HWP) was tested using the exact test of Hardy–Weinberg proportion for multiple alleles21.
Structural analysis of the human SBF1 with different numbers of (GCC)-repeats
We investigated accessibility i.e., probability of being unpaired, of exon 1 of the human SBF1 gene, with 5 to 10 (GCC)-repeats, using the accessibility computation of the ViennaRNA package (RNAplfold with -W 300 -L 300 -u 10)22,23. We compared the accessibilities of all regions of 10 nt length. Furthermore, we used RNAup -b24 to compare possible interactions in homodimeric and heterodimeric SBF1 first exon with different numbers of (GCC)-repeats.
Analysis of the SBF1 (GCC)-repeat across vertebrates
The interval between + 1 and + 100 of the TSS of the SBF1 was searched across all species in which SBF1 was annotated, based on Ensembl 104. The Ensembl alignment program was used for the sequence alignments across the selected species.
Results
The SBF1 (GCC)-repeat allele distribution was significantly different in the NCD group versus controls
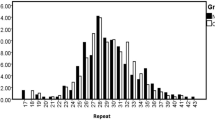
We detected two predominantly abundant alleles of 8 and 9-repeats, which formed > 95% of the allele pool across the two groups (Table 1, Fig. 1). At significantly lower frequencies, we detected repeats of 5, 6, 7, and 10, with frequencies of < 0.03. The allele frequency distribution was significantly different in the NCD group versus controls (Fisher’s exact p = 0.006). Specifically, the frequency ratio of the 8 and 9 repeats was in the reverse order in the NCD group as a result of excess of the 8-repeat in this group.

Allele frequency of the SBF1 (GCC)-repeat in the human samples studied. While multiple alleles were detected, the 8 and 9-repeat alleles were predominantly abundant. Significant excess of the 8-repeat was detected in the NCD group versus controls.
The SBF1 (GCC)-repeat genotype distribution deviated from HWP in both groups and was different between the two groups
The genotype distribution was anomalous in both NCD and control groups, and deviated from the HWP (p < 0.001). Specifically, rather than an expected > 45% 8/9 genotype based on the 8 and 9-repeat allele frequencies, we detected < 18% of that genotype across the two groups (Table 2, Fig. 2). There were other discrepancies in the genotype distribution The 6/8 genotype was significantly more detected than the 6/9 genotype across the human samples studied (Fisher’s exact p = 0.0001).

Genotype frequency of the SBF1 (GCC)-repeat in the human samples studied. The genotype distribution departed from HWP in both groups and was different between the two groups.
The genotype distribution was significantly different between the NCD and control groups (Fisher’s exact p = 0.001) (Table 2), Specifically, we detected significant enrichment of the 8/9 genotype in the NCD group versus controls, and reverse ratio of 8/8 and 9/9 genotypes between the two groups.
Identification of an extreme genotype in the NCD group only
We detected a genotype at the extreme short end of the allele range in one instance of late-onset NCD. This genotype was 5/6 (Fig. 3), and was detected in an 85-year-old female case of NCD with AMTS = 3, and suspected of having late-onset AD. The shortest allele detected in the control group was 6-repeats, and 5-repeats was not detected in this group.

Identification of a genotype at the short extreme of the allele range in one instance of late-onset NCD.
The number of (GCC)-repeats may change the RNA secondary structure and interaction sites
The accessibility of exon 1 of human SBF1 varied with the number of (GCC)-repeats in three regions, around nucleotide (nt) 50 (at the (GCC)-repeat itself), at about nt 200 (at the translation start site) and at nt 220 (all nt relative to the TSS based on Ensembl transcript ID: ENST00000380817.8 SBF1-202) (Fig. 4). Furthermore, we analyzed where the preferred interaction sites would be, and found that there are two different groups of interaction sites (Table 3): in one group, the best molecular interaction occurs between nt 119–130 and nt 219–230, while the other group has interactions between nt 182–200 and nt 193–211.

Accessibility (probability of being unpaired) of all regions of 10 nt length, ending at base x for the first exon of human SBF1 with 5 to 10-repeats. Differences in 3 regions were detected, at about nt 50, about nt 200, and about nt 220.
SBF1 (GGC)-repeat expanded specifically in primates
Across all the vertebrate species studied, the SBF1 (GCC)-repeat specifically expanded beyond 2-repeats in primates (Fig. 5).

Sequence alignment of the SBF1 (GCC)-repeat across selected vertebrate species. The (GCC)-repeat expanded beyond 2-repeats in primates.
Discussion
The primary importance of (GCC)-repeats stems from a possible link between that type of STR and natural selection, mainly for two reasons: Firstly, (GCC)-repeats are specifically enriched in the exons. Secondly, GC-rich sequences are mutation hotspots25, and frequently interrupted by single nucleotide substitutions. The intact occurrence of the SBF1 (GCC)-repeat in primates, and not in any other order, supports selective advantage in this order.
In both NCD and control groups, the genotype distribution significantly departed from HWP. Not only the expected heterozygosity for the observed allele frequencies was dramatically compromised, but also certain heterozygous/heterozygous ratios were biased.
The accumulated homozygosity could not be attributed to the excess of consanguineous marriages in Iran, as excess of homozygosity in consanguineous societies can contribute to between 2 and 11% homozygosity at a given locus26,27. Sampling error is another explanation for the observed genotypes. All samples were collected from the same districts in Iran, and the results were replicated in both groups. Rare primer binding site mutations are known to provoke null alleles in STRs, and lead to false homozygous genotypes28,29,30. In a review by Dakin and Avise, it was reported that whereas null alleles in frequencies typically reported in the literature introduce rather inconsequential biases on average exclusion probabilities, they can introduce substantial errors into empirical assessments of specific mating events by leading to high frequencies of false parentage exclusions31. While the scope of our research was not assessing specific matings, we double-checked 70 random samples across the two groups with alternative primers (Forward: TCAGGGCTTGACGACAGC, Reverse: CTCGACCCTCAGACCCAG), with alternative binding sites to the original primers, and identical PCR conditions to the original primer set, which confirmed our initial genotyping results. It should be noted that this preliminary study needs to be replicated with independent samples by other groups, in order to confirm the results.
A likely hypothesis that may be put forward is that certain heterozygous genotypes might have been selected against in human in the process of evolution. The studied (GCC)-repeat is located in the 5′ UTR, and it may be speculated that the heterodimer RNAs of, for example, 8/9 and 6/9 have a detrimental effect on the downstream events, such as transcript processing and translation. A possible mechanism might be connected to RNA structure and accessibility. Experimental synthetic stem-loop RNAs have been reported to alter the expression of a number of genes in bacteria32. We could show that the accessibility changes with the number of (GCC)-repeats, and can affect at least exon 1. For example, the 6/8 and 6/9 RNA interactions were differentially grouped in groups 1 and 2, respectively (Table 3).
SBF1 is predominantly expressed in the brain and skeletal muscle, and the protein encoded by this gene is a member of the myotubularin family. Myotubularin-related proteins, namely MTMR2, MTMR13/SBF2 and MTMR5/SBF1 are mainly involved in regulating endolysosomal trafficking33 and mitochondrial functioning34. Dysregulation of SBF1 is linked to late-onset NCDs such as AD17, which is also indicated by the observed genotype anomalies in the NCD group versus controls in our study. An isolate instance of an NCD patient harboring a genotype that consisted of extreme short alleles, may be of significance, while random co-occurrence should also be considered as a possibility. The secondary structure and accessibility effect of the 5/6 genotype were dramatically divergent, and the 5-repeat allele length was not detected in the control group. It is possible that low frequency alleles at the extreme ends of the allele distribution curve are subject to negative natural selection8,12,35.
It remains to be clarified how certain heterozygous genotypes might have been selected against at this locus in human. It is also warranted that this STR is sequenced in larger samples and in a spectrum of neurological disorders.
Conclusion
We report indication of a novel biological phenomenon, in which there is significant selection against certain heterozygous genotypes at a STR locus in the human population. We also report different allele and genotype distribution in late-onset NCD versus controls at this locus. In view of the location of the (GCC)-repeat in the 5′ UTR of the SBF1 gene, it is speculated that specific RNA/RNA or DNA/RNA heterodimers may exert effects that are selected against in the course of evolution. It should be noted that this is a pilot study, which needs to be replicated by independent groups and in different samples.
Data availability
The datasets used and analyzed during the current study are included in this published article (Suppl. 1).
Abbreviations
- AMTS:
-
Abbreviated Mental Test Score
- HWP:
-
Hardy–Weinberg principle
- MTMR5:
-
Myotubularin-Related Protein 5
- nt:
-
Nucleotide
- SBF1:
-
SET binding factor 1
- STR:
-
Short tandem repeat
- TSS:
-
Transcription start site
- UTR:
-
Untranslated region
References
Nikkhah, M., Rezazadeh, M., Khorram Khorshid, H. R., Biglarian, A. & Ohadi, M. An exceptionally long CA-repeat in the core promoter of SCGB2B2 links with the evolution of apes and Old World monkeys. Gene 576, 109–114 (2016).
Mohammadparast, S., Bayat, H., Biglarian, A. & Ohadi, M. Exceptional expansion and conservation of a CT-repeat complex in the core promoter of PAXBP1 in primates. Am J Primatol. 76, 747–756 (2014).
Afshar, H. et al. Evolving evidence on a link between the ZMYM3 exceptionally long GA-STR and human cognition. Sci. Rep. 10, 1–7 (2020).
Watts, P. et al. Stabilizing selection on microsatellite allele length at arginine vasopressin 1a receptor and oxytocin receptor loci. Proc. R. Soc. B Biol. Sci. 284, 20171896 (2017).
Hannan, A. J. Tandem repeats mediating genetic plasticity in health and disease. Nat. Rev. Genet. 19, 286–298 (2018).
Ranathunge, C. et al. Microsatellites as agents of adaptive change: An RNA-Seq-based comparative study of transcriptomes from five helianthus species. Symmetry. 13, 933 (2021).
Fotsing, S. F. et al. The impact of short tandem repeat variation on gene expression. Nat Genet. 51, 1652–1659 (2019).
Afshar, H. et al. Natural selection at the NHLH2 core promoter exceptionally long CA-repeat in human and disease-only genotypes in late-onset neurocognitive disorder. Gerontology 66, 514–522 (2020).
Press, M. O., Hall, A. N., Morton, E. A. & Queitsch, C. Substitutions are boring: some arguments about parallel mutations and high mutation rates. Trends Genetics 35, 253–264 (2019).
Press, M. O., Carlson, K. D. & Queitsch, C. The overdue promise of short tandem repeat variation for heritability. Trends Genet. 30, 504–512 (2014).
Annear, D. J. et al. Abundancy of polymorphic CGG repeats in the human genome suggest a broad involvement in neurological disease. Sci. Rep. 11, 2515 (2021).
Jafarian, Z. et al. Natural selection at the RASGEF1C (GGC) repeat in human and divergent genotypes in late-onset neurocognitive disorder. Sci. Rep. 11, 19235 (2021).
Khamse, S. et al. Novel implications of a strictly monomorphic (GCC) repeat in the human PRKACB gene. Sci. Rep. 11, 20629 (2021).
Tang, H. et al. Profiling of short-tandem-repeat disease alleles in 12,632 human whole genomes. Am J Hum Genet. 101, 700–715 (2017).
Namdar, P. et al. Exceptionally long 5′ UTR short tandem repeats specifically linked to primates. Gene 569, 88–94 (2015).
Thierry-Mieg, D. & Thierry-Mieg, J. AceView: A comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 7 Suppl 1, S12.11-14 (2006).
Li, P. et al. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun. 10, 2246 (2019).
Foroughan, M. et al. Validity and reliability of abbreviated mental test score (AMTS) among older Iranian. Psychogeriatrics 17, 460–465 (2017).
Hodkinson, H. M. Evaluation of a mental test score for assessment of mental impairment in the elderly. Age Ageing 1, 233–238 (1972).
Carpenter, C. R. et al. Accuracy of dementia screening instruments in emergency medicine: A diagnostic meta-analysis. Acad Emerg Med. 26, 226–245 (2019).
Guo, S. W. & Thompson, E. A. Performing the exact test of Hardy-Weinberg proportion for multiple alleles. Biometrics 48, 361–372 (1992).
Bernhart, S. H., Mückstein, U. & Hofacker, I. L. RNA accessibility in cubic time. Algorithms Mol. Biol. 6, 3 (2011).
Lorenz, R. et al. ViennaRNA package 2.0. Algorithms Mol. Biol. 6, 26 (2011).
Mückstein, U. et al. Thermodynamics of RNA–RNA binding. Bioinformatics 22, 1177–1182 (2006).
Nesta, A. V., Tafur, D. & Beck, C. R. Hotspots of human mutation. Trends Genet. 37, 717–729 (2021).
Woods, C. G. et al. Quantification of homozygosity in consanguineous individuals with autosomal recessive disease. Am. J. Hum. Genet. 78, 889–896 (2006).
Kirin, M. et al. Genomic runs of homozygosity record population history and consanguinity. PLoS ONE 5, e13996 (2010).
Blais, J. et al. Risk of misdiagnosis due to allele dropout and false-positive PCR artifacts in molecular diagnostics: Analysis of 30,769 genotypes. J Mol Diagn. 17, 505–514 (2015).
Yao, Y. et al. Null alleles and sequence variations at primer binding sites of STR loci within multiplex typing systems. Leg. Med. (Tokyo) 30, 10–13 (2018).
Li, F. et al. Identification of new primer binding site mutations at TH01 and D13S317 loci and determination of their corresponding STR alleles by allele-specific PCR. Forensic Sci. Int. Genet. 8, 143–146 (2014).
Dakin, E. E. & Avise, J. C. Microsatellite null alleles in parentage analysis. Heredity 93, 504–509 (2004).
Paulus, M., Haslbeck, M. & Watzele, M. RNA stem-loop enhanced expression of previously non-expressible genes. Nucleic Acids Res. 32, e78 (2004).
Mammel, A. E. et al. Distinct roles for the Charcot-Marie-Tooth disease-causing endosomal regulators Mtmr5 and Mtmr13 in axon radial sorting and Schwann cell myelination. Hum. Mol. Genet. 31, 1216–1229 (2022).
Berti, B. et al. Bi-allelic variants in MTMR5/SBF1 cause Charcot-Marie-Tooth type 4B3 featuring mitochondrial dysfunction. BMC Med. Genomics 14, 157 (2021).
Khamse, S. et al. Predominant monomorphism of the RIT2 and GPM6B exceptionally long GA blocks in human and enriched divergent alleles in the disease compartment. Genetica 150, 27–40 (2022).
Author information
Authors and Affiliations
Contributions
S.K. and S.A. performed the molecular experiments and allele and genotype analyses. S.H.B. performed the RNA structural and accessibility analyses. H.A. collected the human samples and their clinical information. A.D. contributed to data collection and coordination. M.O. conceived and supervised the project, and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khamse, S., Alizadeh, S., Bernhart, S.H. et al. A (GCC) repeat in SBF1 reveals a novel biological phenomenon in human and links to late onset neurocognitive disorder. Sci Rep 12, 15480 (2022). https://doi.org/10.1038/s41598-022-19878-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-19878-y
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
