
Abstract
The aftermath of traumatization lives on in the neural and epigenetic traces creating a momentum of affliction in the psychological and social realm. Can psychotherapy reorganise these memories through changes in DNA methylation signatures? Using a randomised controlled parallel group design, we examined methylome-wide changes in saliva samples of 84 female former child soldiers from Eastern DR Congo before and six months after Narrative Exposure Therapy. Treatment predicted differentially methylated positions (DMPs) related to ALCAM, RIPOR2, AFAP1 and MOCOS. In addition, treatment associations overlapped at gene level with baseline clinical and social outcomes. Treatment related DMPs are involved in memory formation—the key agent in trauma focused treatments—and enriched for molecular pathways commonly affected by trauma related disorders. Results were partially replicated in an independent sample of 53 female former child soldiers from Northern Uganda. Our results suggest a molecular impact of psychological treatment in women with war-related childhood trauma.
Trial registration: Addressing Heightened Levels of Aggression in Traumatized Offenders With Psychotherapeutic Means (ClinicalTrials.gov Identifier: NCT02992561, 14/12/2016).
Similar content being viewed by others


Introduction
Former child soldiers, particularly female combatants, are at high risk of experiencing multiple, severe and traumatic stressors including violence and repeated rape. These extreme and intense stressors produce lasting changes in mind and body leading to a range of mental disorders. These include posttraumatic stress disorder (PTSD), depression, dissociative symptoms and high levels of aggression against oneself and others1,2. Frequent exposure to severe stressors is decisively detrimental. Such exposure reorganises the functioning of the brain and mind in a lasting, self-perpetuating manner such that simple cues, sometimes arising from imaginative processes alone, may activate a physical response as part of the defence cascade3. Most importantly, the hypothalamic–pituitary–adrenal (HPA) axis may be tilted4,5,6, resulting in anything from a long-lasting immune problem7,8,9 via deficient metabolic systems10,11,12 to the broad trauma related psychopathology maintained by the intrinsic dynamics of traumatic memories.
These processes are realised through epigenetic modifications in neuronal and peripheral tissues, and in particular have been associated with DNA methylation (hereafter DNAm)13,14. For example, PTSD and depression have been associated with DNAm changes in blood and buccal cells at genes involved in glucocorticoid functioning, such as the FKBP prolyl isomerase 5 (FKBP5) and the nuclear receptor subfamily 3 group C member 1 (NR3C1)15,16,17,18,19. Emerging evidence suggests that these associations could even be transferable across generations20,21,22, although studies in mammals are still scarce23. Aggression has also been associated with multiple differentially methylated regions (DMRs) in at least 30 gene promoters24. Likewise, antisocial personality disorder has been associated with DNAm changes in the monoamine oxidase A (MAOA) and B (MAOB)25, two genes relevant in the dopaminergic and serotonergic pathways.
It is yet to be seen whether these epigenetic marks can be modified through treatment such that the clinical symptoms may recede. A few studies with small sample sizes that investigated only a limited number of target genes nonetheless provide early evidence that this may be possible26,27,28. However, so far, there have been no randomised controlled trials using an epigenome-wide association approach to investigate the impact of successful trauma treatment in any sample, let alone in children of war. Given that childhood experiences are important for both aetiology and epigenetic predisposition of mental disorders29,30,31, investigating therapeutic impacts on the epigenome in patients with childhood and war-related trauma may support further development of treatment for mental disorders and contribute to our understanding of therapeutic agents.
We examined a subsample of 84 highly traumatized female former child soldiers who participated in a randomised controlled trial32 using an epigenome-wide approach (EWAS) that investigated associations between differentially methylated positions (DMPs), trauma related mental disorders and social consequences as well as Narrative Exposure Therapy (NET)33 in comparison to a less effective treatment [step 1]. NET is one of the empirically supported psychotherapies for trauma-related disorders34, alongside CPT (Cognitive Processing Therapy), EMDR (Eye Movement Desensitization and Reprocessing) and PE (Prolonged Exposure Therapy). It particularly targets survivors of multiple traumatic stressors. Recent adaptations of NET have successfully treated trauma related suffering, focusing on PTSD, depression and appetitive aggression, and mitigated social problems such as current violent behaviour and perceived social acknowledgment as victim or survivor32,35,36. Then, we identified overlap in associated DMPs [step 2] and further investigated outcome and treatment associations using gene-ontology and gene-network enrichments to determine potential functions [step 3]. Finally, we used an independent cohort of 53 female former child soldiers from Northern Uganda who also had been successfully treated using NET to replicate our findings [step 4].
Results
At baseline, mean Beta per probe ranged from 0.030 to 0.968 with an overall mean value of 0.61 (0.609:0.611; 95% CI). Most probes (N = 305,868) had a methylation status of more than 0.5 (69.7%). Mean DNAm per participant (average Beta over all CpGs) was 0.61 (SD = 0.008). It did not significantly change over time (likelihood-ratio test; LRT = 0.78, p = 0.378) nor did it differ between treatment groups (LRT = 0.14, p = 0.078; Fig. 1A). Although participants in TAU and NET had a similar age of 18 (SD = 1.8) (t = 0.06, df = 81, p = 0.955), epigenetic age differed among groups. In TAU, epigenetic age was larger than reported age at baseline (t = 3.29, df = 41, p = 0.002) and follow-up (t = 3.83, df = 41, p < 0.001), whereas no statistical differences were found in NET (Fig. 1B). For summary statistics see Table 1.

Violin plots showing individual observations of discovery sample. (a) Mean DNAm observed over all estimates (average Beta value) of probes used in EWAS. (b) Mean reported age and mean epigenetic ages. Points represent study participants. Colors represent treatment condition (TAU vs. NET). Assessment time refers to baseline (BL) and 6 month follow-up (FU6). Asterisks mark significant t-test comparisons (***p < 0.001; **p < 0.01).
Step 1: CpGs and genes associated with outcomes and treatment
Linear models (EWAS) showed a varying number of CpGs that were significantly associated (FDR = 0.05) with clinical and social outcomes at baseline. We found significant DNAm associations with PSS-I (DMP at cg02192673; NPFFR2), CVB (51 DMPs), AAGS (DMP at cg20866785; ARHGAP10) and SAQ (85 DMPs), but we did not find significant associations with PHQ-9 or AAS. For details on all estimates see supplement 2.
Patients who received NET showed a significant DNAm reduction at cg23719209 (ALCAM), cg12337669 (AFAP1) and cg08739828 (MOCOS; promoter-linked), and DNAm increase at cg18803039 (RIPOR2) (Fig. 2). Linear model outputs are shown in supplement S2.

Significant associations between treatment condition and DNAm change. Mean DNAm (%) as a function of assessment time at discovery and replication sample (colour coded) for the four CpGs that significantly had an association between treatment effect and DNAm. Dots represent group means and bars show standard errors. Note that samples had different follow-up assessment times (FU4,6,10; numbers represent months) after baseline (BL). Replication sample does not support findings statistically in discovery sample.
Step 2: Intersections of associated CpGs and genes
Significantly associated CpGs and their respective genes were specific to each tested outcome. Only when considering the top 0.1% CpGs ranked by significance (305 probes per association), CpG sites were found to be associated with more than one outcome, with the highest intersection between PSS-I and PHQ-9 (31 CpGs). Six other CpGs were associated both with treatment and any of the mental health measures (for details, see supplement S3). Regarding gene intersections, we found again the highest overlap between PSS-I and PHQ-9 (42 genes). Various other intersections were found among associations. Importantly, a total of 62 genes were associated with treatment and one or more outcome (Fig. 3) (supplement S3).

Intersection plot of genes associated with outcomes and treatment. On the left, intersections are shown as two or more circles connected by lines, whereas single associations are shown as isolated circles at the bottom. On the right, horizontal bars show the total number of intersections and single associations. Intersections with treatment are additionally specified with gene symbols. Treatment is marked in orange. Association sample sizes derive from each of the top 0.1% DMPs identified after EWAS (n = 305 per association).
Step 3: Gene function and network enrichments
Genes associated with treatment and clinical/social outcomes (from top 0.1% CpGs after EWAS) were significantly enriched for multiple networks and functional pathways, after statistical overrepresentation tests in STRING (for details, see supplement S4). We did not find significant enrichment for genes associated with PSS-I. For treatment-related CpGs (PPI enrichment < 0.001), we found a gene-ontology enrichment linked to cortisol and aldosterone synthesis and secretion (strength = 0.88 and 0.77; FDR = 0.015), extracellular matrix (ECM) receptor interaction (strength = 0.77, FDR = 0.016), bile secretion (strength = 0.76, FDR = 0.027), glutamatergic synapse (strength = 0.74, FDR = 0.015), gonadotropin releasing hormone (GnRH) signalling pathway (strength = 0.73, FDR = 0.017). Further enrichment pointed to general body functions, such as nervous system and morphogenesis. Of the seven significantly enriched biological processes, three were associated with embryonic development. When considering the 62 genes intersecting between treatment and outcomes, we further found enrichment for cardiomyopathy (strength = 1.16, FDR = 0.025), oxytocin signalling (strength = 0.93, FDR = 0.043), epidermal growth factors (strength = 0.88, FDR = 0.035) and several biological processes and cellular components (supplement S4).
Step 4: Replication
For ALCAM and AFAP1 we found respectively 4 and 8 CpGs significantly associated with treatment, but these did include cg23719209 or cg12337669 which we found associated in the discovery sample; cg18803039 (RIPOR2) and cg08739828 (MOCOS) were also not successfully replicated. Regarding the treatment vs. outcome association intersections (131 CpGs related to 62 genes), replication was successful for several loci. Eleven out of 64 (17%) CpGs that were associated with treatment in the discovery sample were successfully replicated, namely cg06125671 (ABTB2) and cg22356726 (KIF20B) with the strongest replications (> 1.5% percentage change), followed by cg12774902 (CHRNA10), cg03259703 (IGSF21), cg16662768 (IRF8), cg09102332 (ITGB8), cg15509069 (SDHAF3), cg04070142 (SLCO1A2), cg06665305 (SOWAHC), cg20401896 (WDPCP), cg26212904 (WSCD1) (> 0.5% percentage change). For details, see supplement S5.
Discussion
The present study investigated patterns of DNAm associated with trauma related mental problems and psychotherapeutic treatment in female former child soldiers from Eastern DR Congo. DNAm of different CpGs were associated with treatment but were not replicated in an independent sample. Associated genes found in discovery sample play a critical role in memory formation and fear learning/extinction and, in addition, have been associated with processes of the immune system. Moreover, 62 genes were related to both clinical/social outcomes and treatment; replication was successful in 17% of the tested CpGs. Enrichment analyses corroborated that NET might alter gene regulatory processes involved in memory formation (ECM interaction, focal adhesion, glutaminergic signalling) along with alterations of the glucocorticoid and aldosterone system amongst others. Interestingly, genes involved in GnRH signalling were associated with both social acknowledgement and treatment.
Effect of NET treatment on DNAm
Using a longitudinal parallel group design to draw causal effects of NET vs. TAU on DNAm change, we found that treatment significantly predicted DNAm change from baseline to six month follow up in four CpGs (ALCAM, AFAP1, MOCOS and RIPOR2). Activated leukocyte cell adhesion molecule (ALCAM) and actin filament associated protein 1 (AFAP1) were replicated, despite the limited power of the replication sample. Products of both genes have previously been associated with the formation of memory. In NET, restructuring of the memory and particularly the establishment of hypersalient/traumatic memories in ‘time and space’ of the autobiographic memory has been postulated as cardinal agent of change for the treatment of PTSD and appetitive aggression33,36,37. Substantial evidence corroborates the involvement of actin filaments in neural plasticity, specifically the generation, stabilisation and consolidation of long-term memory38. Actin dynamics interact with several of the identified enriched gene networks, namely glutaminergic signalling, focal adhesion, ECM-receptor interaction (see review by Rudy39) as well as the RAP1 signalling pathway40. ALCAM is a member of the immunoglobulin superfamily and important for the growth of midbrain dopamine neurons through trans-heterophilic interactions41,42. Dopamine neurons in the midbrain are crucial for reinforcement learning (see review by Daw et al.43). Actin polymerization and midbrain dopamine neurons have both been found to modulate fear extinction in animal studies44,45,46,47. ALCAM was furthermore linked to maintaining the integrity of the blood–brain-barrier and the proliferation of T-cells48,49. In line with this, NET has previously been found to normalise regulatory T cells50, which have been reported to be reduced in individuals with PTSD51.
Further, among the top 0.1% significant CpGs (n = 305), loci on 62 genes were associated both with the effect of treatment and clinical/social outcomes in the discovery sample and 11 (17%) were replicated in the independent sample (percentage change of 0.5% on the same CpG in the same direction). Percentage change in the replication sample was strongest in ABTB2 and KIF20B. The former encodes the ankyrin repeat and BTB/POZ domain-containing protein 2, which inhibits the aggregation of alpha-synuclein and exerts a protective effect on dopamine neurons usually killed in Parkinson disease52. The second encodes a kinesin-like protein relevant to cytokinesis during cell division53,54. Moreover, CHRNA10 encodes neuronal nicotinic acetylcholine receptor (nAChR) complex α9 and α10, and its inhibition has recently been suggested to prevent neuropathic pain55. Deletion of nAChR α9 also resulted in altered bone structure in mice56. Other genes have been related to the immune system (IGSF21, IRF8, SOWAHC), bioenergetics (SDHAF3) and integrins (ITGB8).
Finally, epigenetic age is expected to be related to disease and mental health57. Increasing evidence suggests that biological, social and environmental factors induce an accelerated epigenetic aging58. Unfortunately, we did not find conclusive results regarding the effect of treatment on epigenetic age, since our groups had differing baseline values and there were no differences in epigenetic age between timepoints. More research is also needed in that direction.
Gene ontology analysis
Gene ontology analysis provided further insights into biological mechanisms underlying the changes triggered by NET. Seven out of 63 genes involved in cortisol synthesis and secretion have been found associated with treatment in our sample, indicating changes in HPA-regulation, i.e., alterations in the elite force of defence5. Additionally, multiple genes associated with treatment are involved in aldosterone syntheses and secretion. Aldosterone is a mineralocorticoid steroid hormone that is involved in the renin–angiotensin–aldosterone-system (RAAS), crucial for the homeostatic regulation of body fluids and electrolytes. It is also implicated in cardiovascular and metabolic diseases, by mediating immune cell activation59. Elevated aldosterone levels were previously found in patients with depression and PTSD60,61,62. Glutaminergic signalling is another pathway related to treatment in our sample. Most cortical synapses in the human brain are glutamatergic (80–90%). Alteration of glutaminergic signalling is essential in the development of trauma related psychiatric disorders, which motivated the investigation into a variety of therapeutics, but with inconsistent outputs63. Further pathways that currently emerged in association with the treatment have traditionally been related to social behaviour. For example, oxytocin signalling has been described to mediate complex social behaviours in animal and human studies64. Moreover, GnRH was related with both social acknowledgement and treatment. GnRH is synthesised and released by GnRH neurons in the hypothalamus, especially during puberty and adolescence.
DNAm markers associated with clinical and social outcomes
Our analysis revealed a mixture of overlapping epigenetic signatures associated with clinical and social outcomes (Fig. 3). This result may be explained by the fact that psychological outcomes share commonalities and are intrinsically correlated with each other, although addressing distinct mental constructs. Significant associations were found for perceived social acknowledgement as victim or survivor, current violent behaviour, PTSD severity and guilt, but not for depression or AAS. Identified genes are involved in HPA axis activation and pain modulation (NPFFR2)65,66, neural functioning (SLC2A4 and KCNG2)67,68 immune system (TRIM59; ref: UniProtKB Q8IWR1), chromatic remodelling (UBL4A)69, insulin metabolism (SLC2A4)70, and transcriptional fine tuning in the early post-natal period (DNMT3A)71,72, amongst others. The multiple DMPs (i.e., on promoters) associated with social acknowledgement indicate a potential sensitivity of DNAm for processes that originate in the social environment.
Is DNA methylation associated with mental disorders reversible?
If we assume that complex traits, such as trauma related disorders are in part influenced by epigenetic markers, one might expect that treating mental disorders reverses epigenetic changes in specific loci responsible for the development of symptoms. Our data, however, does not support this hypothesis. It is noteworthy that almost none of the CpGs associated with trauma related disorders were also associated with treatment, despite various intersections at the gene level. This indicates that treatment does not promote a simple reversal of epigenetic alterations on specific CpG sites that correlate with trauma related disorder symptoms. Instead, an adaptation of regulatory routes that are pathological or render survivors socially dysfunctional might contribute to psychological and physiological changes in response to treatment. Additionally, it is still unclear whether and to which degree methylation changes could effectively produce meaningful biological impacts in the context of psychotherapy. Research investigating biomarkers for trauma related disorders are still incipient, compared to substance use73, smoking74 or cancer75. Considering that our study detected small levels of DNAm change after treatment application, our findings should be interpreted with caution. Further studies should also test the hypothesis that therapy treats phenotypic conditions through alternative epigenetic routes rather than by reversing specific methylated positions on the genome.
Limitations
Although our study has several strengths, the results need to be interpreted in light of the following limitations. Firstly, we cannot be sure that the associations found in our study directly reflect gene expression and protein levels in participants, although a growing body of research suggests that DNAm is a reliable epigenetic marker for gene regulation76. Secondly, the lack of significant CpG sites associated with PTSD and appetitive aggression might be due to the low variance in these particular phenotypes, since participants were preselected based on presence of a PTSD diagnosis and apparent aggressive behaviour. Thirdly, we should acknowledge that individual measures of DNAm might have been affected by aspects outside of the control of our design, such as menstrual cycle, contact with chemical substances or diet. However, systematic bias should be reduced by the randomized study design. Also, although the estimation and processing of methylation data in our study was done meticulously, we did not confirm it using a validation technique such as pyrosequencing. Further, we did not correct for multiple testing at the level of psychological outcomes, which could reduce the number of significant associations. Also, our replication method supported the association of some DMPs, but the magnitude of change was relatively small, which may mean there was not an effective change in methylation status after 6–9 months of treatment application. As indicated above, methylation change might qualitatively distinguish treatment groups, but the degree of change has still an uncertain meaning. Lastly, we cannot rule out tissue-specificity and genetic population stratification in our study. Genetic restriction should not be mistakenly understood as genetic bias however. Since we applied a paired sample design, baseline versus follow-up, individuals were their own genetic controls.
In conclusion, this study is the first to suggest that evidence based trauma therapy—specifically Narrative Exposure Therapy (NET)—might affect DNAm markers in multiple genes associated with psychiatric conditions such as PTSD, but also with trauma related social problems such as current violent behaviour and social acknowledgement. We found evidence that NET might effectively alter DNAm of loci close to genes of general and specific biological processes and pathways cardinal to trauma related problems and somatic complaints as well as social behaviour and reproduction.
Methods
Study procedure and participants
We applied a prospective randomised parallel group design in a sample of 88 female former child soldiers in the Eastern DR Congo. All participants fulfilled the DSM-5 diagnostic criteria for PTSD. Inclusion criteria were female sex, minimum age of 16 years and armed group involvement. Recruited through a local non-profit organisation, patients were invited to meet with psychological interviewers, where the study was outlined in detail and written informed consent was obtained. Simple randomisation was applied to eligible women for allocation to Narrative Exposure Therapy (NET) or treatment-as-usual (TAU; see Robjant et al.32) condition via SPSS by a person who was not involved in interviews or treatment. Four women did not offer saliva samples and were thus excluded from this analysis. The final sample included 84 women (nnet and ntau = 42) with saliva samples collected at baseline (BL) and 6-month follow-ups (FU6). Participants had a mean age of 18 (SD = 1.9) years at baseline. All women had a history of abduction and numerous lifetime traumatic events77. It is very uncommon to smoke tobacco within this micro-culture, and other drugs were not available or affordable for these women, therefore this information was not gathered during interviews. All methods were performed in accordance with the relevant guidelines and regulations.
Assessment
Structured psychodiagnostic interviews were conducted by trained local interviewers blind to treatment condition. Baseline assessment included (1) sociodemographic questions, (2) a 44-items checklist for traumatic events and perpetrated acts of violence, and (3) clinical and social outcomes. Clinical outcomes were PTSD (PSS-I; DSM-5 Posttraumatic Stress Symptom Scale Interview78), depression (PHQ-9; Patient Health Questionnaire79) and appetitive aggression (AAS; Appetitive Aggression Scale80). Social outcomes were current violent behaviour (CVB; checklist of Current Violent Behaviour against children, intimate partner, and others81), feelings of guilt (AAGS, Attitudes About Guilt Survey82) and perceived recognition of the trauma and support in the women’s social environment (SAQ; Social Acknowledgement Scale83). A more detailed description of the sample, study design, questionnaire measures and treatment is provided in Robjant et al.32.
Saliva samples were collected at baseline and 6-month follow-up using Oragene DNA OG-500 kits (DNA Genotek, Ontario, Canada), which efficiently preserve DNA material at room temperature and above even over long periods of time84. Samples were stored in dry carton boxes in Goma, DR Congo, before DNA extraction and DNAm profiling (GenomeScan, Leiden, Netherlands). DNA was purified using QIAsymphony DSP DNA Midi Kit (Qiagen, Hilden, Germany) and concentrations were determined using Invitrogen Quant-iT™ PicoGreen™ dsDNA Assay Kit (Thermo Fisher Scientific, USA). Bisulfite conversion using 500 ng of genomic DNA was performed using EZ DNA Methylation Gold Kit (Zymo Research, USA), and methylation profiling was conducted using the Infinium MethylationEPIC BeadChip array (Illumina, California, USA). There were no deviations from the Illumina protocol and all experiments were performed in compliance with GenomeScan Standard Operating Procedures (SOPs). Quality control was carried out using Illumina Technical Controls Plots and using the MethylAid R package85. Less than 16 months elapsed between saliva collection and biochemical analysis. Raw methylation data containing 865,859 probes was processed to remove probes of bad quality (n = 12,933), non CpG or SNPs probes (n = 29,873), probes at male sex chromosomes (n = 58) and cross-reactive probes (n = 124,489). The remaining 698,540 probes were filtered to select differentially methylated positions (DMPs). In total, 305,868 CpGs were selected for statistical analyses. During processing of methylation data, we accounted for batch effects and generated estimates of epigenetic age86, sex (all participants were confirmed as female), and cell counts87 (see supplement S1) (Fig. 4).

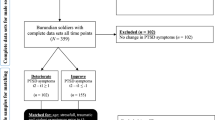
Scheme of study methodology. From top to bottom, diagram shows the four main steps of data analysis, after preliminary data collection and processing. Step 1: epigenome-wide association analysis to identify biomarkers for clinical and social outcomes considering baseline data only. Treatment association considered DNAm change accounting for baseline and follow-up data. Step 2: extraction of CpGs and genes from identified associations and test for intersections. Step 3: gene-ontology and gene-network analysis was conducted for those associations where treatment and other outcomes had overlapping results to infer relevant body functions. Step 4: replication of results in an independent sample using linear models and observing directionality and intensity of methylation change.
Data analysis
To investigate general differences in overall DNAm between treatment groups, we conducted a generalized linear mixed model using the formula DNAm ~ treatment + time + treatment × time + reported age + nr. traumatic events + cell counts, where response was mean Beta across all CpGs. Control variables were set to account for individual differences, whereas participant IDs were set as random effects. Epigenetic age was compared among groups using t-tests.
To investigate the effect of treatment on DNAm, we performed an analysis in four steps: [1] EWAS: an epigenome wide association study (EWAS) approach to find DNAm associations with outcomes and treatment by fitting robust generalized linear models using the formula Y ~ X + reported age + nr. of lifetime traumatic events + cell counts. We chose a robust model to account for potential violation of statistical assumptions. For outcomes, Y was the M-value (transformation of Beta values to normalize data) at baseline for each probe, and X was one of the outcomes. We fitted these model structures for each outcome separately to avoid multicollinearity. For treatment, Y was the Beta value difference between FU6 and BL, X was treatment condition, and cell counts were the mean cell count values between BL and FU6. Reported age, number of lifetime traumatic events (perpetrated and experienced) and cell counts were treated as covariates to control for individual differences and past experiences. P-values were adjusted for multiple testing after Benjamini-Hochberg88 and used to rank probes by significance (cut-off: FDR = 0.05) per outcome. QQ and MD plots were used to evaluate model fits. Statistical models were implemented in lme489 and limma90, using R 3.5.191 in RStudio92. [2] Probe annotation, CpG/gene intersections: first, metadata for probes were gathered using minfi93 and complemented with the updated Illumina EPIC array annotation release 03.13.201794. CpGs that did not correspond to a specific gene were manually investigated using the UCSC genome browser (GRCh37/hg19 assembly), and the closest protein-coding gene was selected. All gene symbols were double-checked against the most recent HUGO (Human Genome Organization) nomenclature. Second, to find biomarkers in our sample that were both associated with clinical/social outcomes and with treatment, we intersected CpGs and genes associated with those variables. For this, we selected the top 0.1% CpGs (305 probes) ordered by adjusted p-value for each variable. Contrary to a conservative FDR cut-off, this allowed us to inspect a more comparable number of sets per association. [3] Gene network analysis: to investigate gene function and important biological pathways, we conducted a gene-network and gene-ontology analysis using STRING95. [4] Replication: statistical analyses were replicated in a subsample of female former child soldiers abducted by the Lord’s Resistance Army in Northern Uganda of whom all received NET (N = 53). These participants had a mean age of 34 (SD = 9.1) years and a history of trauma and perpetration comparable to the discovery sample. Phenotype and saliva samples were collected at baseline, 4-month (FU4) and 10-month (FU10) follow-up interviews. Phenotypic data and DNAm processing are described elsewhere96,97. Baseline biomarker correlates were investigated for clinical outcomes only: PTSD (PDS subscale employed as a structured interview98), depression (HSCL99) and appetitive aggression (AAS). To also control for trauma load, we used a trauma checklist specific for the Ugandan context, where information about violence exposure and perpetration was present100. Linear models were calculated for those CpG sites found in associations within the discovery sample by using the formula and procedure mentioned above. Replication was considered successful for the effect of treatment when the direction of DNAm change was identical with a minimum percentage change of 0.5% in both discovery and replication samples. Average percentage change in CpG methylation across all CpG sites for the entire discovery sample was 0.26% (SD = 0.89).
Statement of ethics
All participants have given written informed consent. Concerning the discovery sample, the study protocol was approved by the Ethical Commission of the University of Konstanz and the Social Funds of the DR Congo. Concerning the replication sample, the study protocol was approved by the Ethical Commission of the University of Konstanz, the Gulu University Research and Ethics Committee (Uganda), the Lacor Hospital Institutional Research Ethics Committee (Uganda), and the Ugandan National Council of Science and Technology.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Schauer, E. & Elbert, T. In Trauma Rehabilitation After War and Conflict: Community and Individual Perspectives (ed. Erin, M.) 311–360 (Springer, 2010).
Betancourt, T. S. et al. Research review: Psychosocial adjustment and mental health in former child soldiers—a systematic review of the literature and recommendations for future research. J. Child Psychol. Psychiatry 54, 17–36 (2013).
Schauer, M. & Elbert, T. Dissociation following traumatic stress—etiology and treatment. Zeitschrift für Psychologie / J. Psychol. 218, 109–127. https://doi.org/10.1027/0044-3409/a000018 (2010).
Elbert, T., Weierstall, R. & Schauer, M. Fascination violence: On mind and brain of man hunters. Eur. Arch. Psychiatry Clin. Neurosci. 260, 100–105 (2010).
McEwen, B. S. & Lasley, E. N. The End of Stress as We Know It (Joseph Henry Press, 2002).
Yehuda, R. et al. Post-traumatic stress disorder. Nat. Rev. Dis. Primers. 1, 1–22 (2015).
Gola, H. et al. Posttraumatic stress disorder is associated with an enhanced spontaneous production of pro-inflammatory cytokines by peripheral blood mononuclear cells. BMC Psychiatry 13, 40 (2013).
Geiger, M. L. et al. Investigating the effects of childhood maltreatment on pro-inflammatory signaling: The influence of cortisol and DHEA on cytokine secretion ex vivo. Mental Health Prevent. 13, 176–186 (2019).
Boeck, C. et al. Inflammation in adult women with a history of child maltreatment: The involvement of mitochondrial alterations and oxidative stress. Mitochondrion 30, 197–207 (2016).
Karabatsiakis, A. et al. Metabolite profiling in posttraumatic stress disorder. J. Mol. Psychiatry 3, 2 (2015).
Mellon, S. H., Gautam, A., Hammamieh, R., Jett, M. & Wolkowitz, O. M. Metabolism, metabolomics, and inflammation in posttraumatic stress disorder. Biol. Psychiat. 83, 866–875 (2018).
Koenig, A. M. et al. Serum profile changes in postpartum women with a history of childhood maltreatment: A combined metabolite and lipid fingerprinting study. Sci. Rep. 8, 1–10 (2018).
Ryan, J., Chaudieu, I., Ancelin, M.-L. & Saffery, R. Biological underpinnings of trauma and post-traumatic stress disorder: Focusing on genetics and epigenetics. Epigenomics 8, 1553–1569 (2016).
Klengel, T. & Binder, E. B. Epigenetics of stress-related psychiatric disorders and gene × environment interactions. Neuron 86, 1343–1357 (2015).
Vinkers, C. H. et al. Traumatic stress and human DNA methylation: A critical review. Epigenomics 7, 593–608 (2015).
O’Donnell, K. J. & Meaney, M. J. Epigenetics, development, and psychopathology. Annu. Rev. Clin. Psychol. 16, 327–350 (2020).
Blacker, C. J., Frye, M. A., Morava, E., Kozicz, T. & Veldic, M. A review of epigenetics of PTSD in comorbid psychiatric conditions. Genes 10, 140 (2019).
Vukojevic, V. et al. Epigenetic modification of the glucocorticoid receptor gene is linked to traumatic memory and post-traumatic stress disorder risk in genocide survivors. J. Neurosci. 34, 10274–10284 (2014).
Ramo-Fernández, L. et al. The effects of childhood maltreatment on epigenetic regulation of stress-response associated genes: An intergenerational approach. Sci. Rep. 9, 1–12 (2019).
Serpeloni, F. et al. Grandmaternal stress during pregnancy and DNA methylation of the third generation: An epigenome-wide association study. Transl. Psychiatry 7, e1202. https://doi.org/10.1038/tp.2017.153 (2017).
Serpeloni, F. et al. Does prenatal stress shape postnatal resilience? An epigenome-wide study on violence and mental health in humans. Front. Genet. 10, 269. https://doi.org/10.3389/fgene.2019.00269 (2019).
Braithwaite, E., Kundakovic, M., Ramchandani, P., Murphy, S. & Champagne, F. Maternal prenatal depressive symptoms predict infant NR3C1 1F and BDNF IV DNA methylation. Epigenetics 10, 408–417 (2015).
Horsthemke, B. A critical view on transgenerational epigenetic inheritance in humans. Nat. Commun. 9, 1–4 (2018).
Guillemin, C. et al. DNA methylation signature of childhood chronic physical aggression in T cells of both men and women. PLoS ONE 9, e86822 (2014).
Ziegler, C. & Domschke, K. Epigenetic signature of MAOA and MAOB genes in mental disorders. J. Neural Transm. 125, 1581–1588 (2018).
Yehuda, R. et al. Epigenetic biomarkers as predictors and correlates of symptom improvement following psychotherapy in combat veterans with PTSD. Front. Psych. 4, 118 (2013).
Vinkers, C. H. et al. Successful treatment of post-traumatic stress disorder reverses DNA methylation marks. Mol. Psychiatry 2, 1–8 (2019).
Xulu, K. R. et al. DNA methylation and psychotherapy response in trauma-exposed men with appetitive aggression. Psychiatry Res. 2, 113608 (2020).
Labonte, B. et al. Differential glucocorticoid receptor exon 1B, 1C, and 1H expression and methylation in suicide completers with a history of childhood abuse. Biol. Psychiat. 72, 41–48 (2012).
Jaworska-Andryszewska, P. & Rybakowski, J. K. Childhood trauma in mood disorders: Neurobiological mechanisms and implications for treatment. Pharmacol. Rep. 71, 112–120 (2019).
McGowan, P. O. et al. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 12, 342–348 (2009).
Robjant, K. et al. The treatment of posttraumatic stress symptoms and aggression in female former child soldiers using adapted Narrative Exposure therapy—a RCT in Eastern Democratic Republic of Congo. Behav. Res. Therapy 123, 103482 (2019).
Schauer, M., Neuner, F. & Elbert, T. Narrative Exposure Therapy: A Short-Term Treatment for Traumatic Stress Disorders (Hogrefe Publishing, 2011).
Schnyder, U. et al. Psychotherapies for PTSD: What do they have in common?. Eur. J. Psychotraumatol. 6, 28186 (2015).
Hinsberger, M. et al. Long-term effects of psychotherapy in a context of continuous community and gang violence: changes in aggressive attitude in high-risk South African adolescents. Behav. Cogn. Psychother. 48, 1–13 (2020).
Elbert, T., Hermenau, K., Hecker, T., Weierstall, R. & Schauer, M. In Interventionen bei Gewalt- und Sexualstraftätern: Risko-Management, Methoden und Konzepte der forensischen Therapie (eds Endras, J. et al.) 255–276 (Medizinisch Wissenschaftliche Verlagsgesellschaft, 2012).
Elbert, T., Schauer, M. & Neuner, F. Evidence Based Treatments for Trauma-Related Psychological Disorders 229–253 (Springer, 2015).
Lynch, G., Rex, C. S. & Gall, C. M. LTP consolidation: Substrates, explanatory power, and functional significance. Neuropharmacology 52, 12–23 (2007).
Rudy, J. W. Actin dynamics and the evolution of the memory trace. Brain Res. 1621, 17–28 (2015).
Mun, H. & Jeon, T. J. Regulation of actin cytoskeleton by Rap1 binding to RacGEF1. Mol. Cells 34, 71–76 (2012).
Bye, C. R., Jönsson, M. E., Björklund, A., Parish, C. L. & Thompson, L. H. Transcriptome analysis reveals transmembrane targets on transplantable midbrain dopamine progenitors. Proc. Natl. Acad. Sci. 112, E1946–E1955 (2015).
Bye, C. R., Rytova, V., Alsanie, W. F., Parish, C. L. & Thompson, L. H. Axonal growth of midbrain dopamine neurons is modulated by the cell adhesion molecule ALCAM through trans-heterophilic interactions with L1cam, Chl1, and semaphorins. J. Neurosci. 39, 6656–6667 (2019).
Daw, N. D. & Shohamy, D. The cognitive neuroscience of motivation and learning. Soc. Cogn. 26, 593–620 (2008).
Menezes, J. et al. Facilitation of fear extinction by novelty depends on dopamine acting on D1-subtype dopamine receptors in hippocampus. Proc. Natl. Acad. Sci. 112, E1652–E1658 (2015).
Cai, L. X. et al. Distinct signals in medial and lateral VTA dopamine neurons modulate fear extinction at different times. Elife 9, e54936 (2020).
Mantzur, L., Joels, G. & Lamprecht, R. Actin polymerization in lateral amygdala is essential for fear memory formation. Neurobiol. Learn. Mem. 91, 85–88 (2009).
Motanis, H. & Maroun, M. Differential involvement of protein synthesis and actin rearrangement in the reacquisition of contextual fear conditioning. Hippocampus 22, 494–500 (2012).
Lécuyer, M.-A. et al. Dual role of ALCAM in neuroinflammation and blood–brain barrier homeostasis. Proc. Natl. Acad. Sci. 114, E524–E533 (2017).
Zimmerman, A. W. et al. Long-term engagement of CD6 and ALCAM is essential for T-cell proliferation induced by dendritic cells. Blood 107, 3212–3220 (2006).
Morath, J. et al. The effect of trauma-focused therapy on the altered T cell distribution in individuals with PTSD: Evidence from a randomized controlled trial. J. Psychiatr. Res. 54, 1–10 (2014).
Sommershof, A. et al. Substantial reduction of naive and regulatory T cells following traumatic stress. Brain Behav. Immun. 23, 1117–1124 (2009).
Roy, A. & Pahan, K. Ankyrin repeat and BTB/POZ domain containing protein-2 inhibits the aggregation of alpha-synuclein: Implications for Parkinson’s disease. FEBS Lett. 587, 3567–3574 (2013).
Janisch, K. M., McNeely, K. C., Dardick, J. M., Lim, S. H. & Dwyer, N. D. Kinesin-6 KIF20B is required for efficient cytokinetic furrowing and timely abscission in human cells. Mol. Biol. Cell 29, 166–179 (2018).
Janisch, K. M. et al. The vertebrate-specific Kinesin-6, Kif20b, is required for normal cytokinesis of polarized cortical stem cells and cerebral cortex size. Development 140, 4672–4682 (2013).
Romero, H. K. et al. Inhibition of α9α10 nicotinic acetylcholine receptors prevents chemotherapy-induced neuropathic pain. Proc. Natl. Acad. Sci. 114, E1825–E1832 (2017).
Baumann, L. et al. Deletion of nicotinic acetylcholine receptor alpha9 in mice resulted in altered bone structure. Bone 120, 285–296 (2019).
Fransquet, P. D., Wrigglesworth, J., Woods, R. L., Ernst, M. E. & Ryan, J. The epigenetic clock as a predictor of disease and mortality risk: A systematic review and meta-analysis. Clin. Epigenetics 11, 1–17 (2019).
Oblak, L., van der Zaag, J., Higgins-Chen, A. T., Levine, M. E. & Boks, M. P. A systematic review of biological, social and environmental factors associated with epigenetic clock acceleration. Ageing Res. Rev. 2, 101348 (2021).
Ferreira, N. S., Tostes, R. C., Paradis, P. & Schiffrin, E. L. Aldosterone, inflammation, immune system, and hypertension. Am. J. Hypertens. 2, 2 (2020).
Emanuele, E., Geroldi, D., Minoretti, P., Coen, E. & Politi, P. Increased plasma aldosterone in patients with clinical depression. Arch. Med. Res. 36, 544–548 (2005).
Murck, H. et al. The renin-angiotensin-aldosterone system in patients with depression compared to controls—a sleep endocrine study. BMC Psychiatry 3, 15 (2003).
Terock, J. et al. Differential activation of the renin-angiotensin-aldosterone-system in response to childhood and adulthood trauma. Psychoneuroendocrinology 107, 232–240 (2019).
Averill, L. A. et al. Glutamate dysregulation and glutamatergic therapeutics for PTSD: Evidence from human studies. Neurosci. Lett. 649, 147–155 (2017).
Churchland, P. S. & Winkielman, P. Modulating social behavior with oxytocin: How does it work? What does it mean?. Horm. Behav. 61, 392–399 (2012).
Elshourbagy, N. A. et al. Receptor for the pain modulatory neuropeptides FF and AF is an orphan G protein-coupled receptor. J. Biol. Chem. 275, 25965–25971 (2000).
Lin, Y.-T. et al. NPFFR2 activates the HPA axis and induces anxiogenic effects in rodents. Int. J. Mol. Sci. 18, 1810. https://doi.org/10.3390/ijms18081810 (2017).
Zhu, X. R., Netzer, R., Böhlke, K., Liu, Q. & Pongs, O. Structural and functional characterization of Kv6.2 a new gamma-subunit of voltage-gated potassium channel. Receptors Channels 6, 337–350 (1999).
Inatome, R. et al. Identification of CRAM, a novel unc-33 gene family protein that associates with CRMP3 and protein-tyrosine kinase(s) in the developing rat brain. J. Biol. Chem. 275, 27291–27302. https://doi.org/10.1074/jbc.m910126199 (2000).
Groothuis, T. A., Dantuma, N. P., Neefjes, J. & Salomons, F. A. Ubiquitin crosstalk connecting cellular processes. Cell Div. 1, 1–7 (2006).
Bowman, P. R. T., Smith, G. L. & Gould, G. W. GLUT4 expression and glucose transport in human induced pluripotent stem cell-derived cardiomyocytes. PLoS ONE 14, e0217885. https://doi.org/10.1371/journal.pone.0217885 (2019).
Kaneda, M. et al. Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429, 900–903 (2004).
Stroud, H. et al. Early-life gene expression in neurons modulates lasting epigenetic states. Cell 171, 1151–1164 (2017).
Andersen, A. M., Dogan, M. V., Beach, S. R. & Philibert, R. A. Current and future prospects for epigenetic biomarkers of substance use disorders. Genes 6, 991–1022 (2015).
Philibert, R. et al. Reversion of AHRR demethylation is a quantitative biomarker of smoking cessation. Front. Psych. 7, 55 (2016).
Mikeska, T. & Craig, J. M. DNA methylation biomarkers: Cancer and beyond. Genes 5, 821–864 (2014).
Suzuki, M. M. & Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 9, 465–476 (2008).
Robjant, K. et al. Trauma, aggression, and post conflict perpetration of community violence in female former child soldiers—A study in eastern DR congo. Front. Psychiatry https://doi.org/10.3389/fpsyt.2020.533357 (2020).
Foa, E. & Capaldi, S. Manual for the administration and scoring of the PTSD symptom scale–interview for DSM-5 (PSS-I-5). (2013).
Kroenke, K. & Spitzer, R. L. The PHQ-9: A new depression diagnostic and severity measure. Psychiatr. Ann. 32, 509–515 (2002).
Weierstall, R. & Elbert, T. The appetitive aggression scale—development of an instrument for the assessment of human’s attraction to violence. Eur. J. Psychotraumatol. 2, 8430 (2011).
Crombach, A. & Elbert, T. Controlling offensive behavior using narrative exposure therapy: A randomized controlled trial of former street children. Clin. Psychol. Sci. 3, 270–282 (2015).
Kubany, E. S. et al. Initial examination of a multidimensional model of trauma-related guilt: Applications to combat veterans and battered women. J. Psychopathol. Behav. Assess. 17, 353–376 (1995).
Maercker, A. & Müller, J. Social acknowledgment as a victim or survivor: A scale to measure a recovery factor of PTSD. J. Trauma. Stress 17, 345–351 (2004).
Burrows, A. M., Kasu, M. & D’Amato, M. E. Preservation of DNA integrity in biological material. Forens. Sci. Int. Genet. Suppl. Ser. 7, 416–418 (2019).
van Iterson, M. et al. MethylAid: visual and interactive quality control of large Illumina 450k datasets. Bioinformatics 30, 3435–3437 (2014).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, 3156 (2013).
Suderman, M., Hemani, G. & Min, J. meffil: efficient algorithms for DNA methylation. R package version 1.1.1. https://github.com/perishky/meffil. (2020).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 2, 289–300 (1995).
lme4: mixed-effects modeling with R (Berlin, 2010).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
R: a language and environment for statistical computing (R Foundation for Statistical Computing, Vienna, Austria, 2018).
RStudio: integrated development for R. RStudio, Inc., Boston, MA. URL: https://www.rstudio.com (2019).
Aryee, M. J. et al. minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369. https://doi.org/10.1093/bioinformatics/btu049 (2014).
Zhou, W., Laird, P. W. & Shen, H. Comprehensive characterization, annotation and innovative use of Infinium DNA methylation BeadChip probes. Nucleic Acids Res. 45, e22–e22 (2017).
Szklarczyk, D. et al. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613 (2019).
Schneider, A. et al. Does cumulative exposure to traumatic stressors predict treatment outcome of community-implemented exposure-based therapy for PTSD?. Eur. J. Psychotraumatol. 11, 1789323 (2020).
Vukojevic, V. et al. Evolutionary conserved role of neural cell adhesion molecule-1 in memory. Transl. Psychiatry 10, 1–13 (2020).
Foa, E. B. et al. Psychometric properties of the posttraumatic diagnostic scale for DSM–5 (PDS–5). Psychol. Assess. 28, 1166 (2016).
Derogatis, L. R., Lipman, R. S., Rickels, K., Uhlenhuth, E. H. & Covi, L. In Psychological Measurements in Psychopharmacology Vol. 7 (eds Pichot, P. & Olivier-Martin, R.) 79–110 (Karger, 1974).
Wilker, S. et al. How to quantify exposure to traumatic stress? Reliability and predictive validity of measures for cumulative trauma exposure in a post-conflict population. Eur. J. Psychotraumatol. 6, 28306 (2015).
Acknowledgements
The authors are grateful for the assistance of the organization PAMI and the teams of therapists and interviewers. We thank the advice of Prof Dr Domenic de Quervain on the first version of the manuscript. We acknowledge the support of the state of Baden-Württemberg (Germany) by providing bwHPC. Prof Dr Thomas Elbert and Anja C. Zeller (scholarship) were supported by the Hector Fellow Academy.
Funding
Open Access funding enabled and organized by Projekt DEAL. The research was funded by the University of Konstanz, and vivo international e.V. supported the project by providing clinical supervision.
Author information
Authors and Affiliations
Contributions
A.K., T.E. and K.R. designed the field study. K.R. coordinated data assessment, treatment delivery and clinical supervision. S.C. conducted discovery data pre-processing (supported by E.U. and D.N.), data analysis and preparation of illustrations as well as replication. Replication data was produced and processed by S.W. and V.V. S.W. and I.T.K. were responsible for data collection of the Replication sample. A.K. provided clinical insights into the epigenetic findings. S.C. and A.K. drafted the manuscript. D.N., V.V., E.U., T.E., S.W., I.T.K. and A.C.Z. critically reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Carleial, S., Nätt, D., Unternährer, E. et al. DNA methylation changes following narrative exposure therapy in a randomized controlled trial with female former child soldiers. Sci Rep 11, 18493 (2021). https://doi.org/10.1038/s41598-021-98067-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-98067-9
This article is cited by
-
Epigenetic impact of a 1-week intensive multimodal group program for adolescents with multiple adverse childhood experiences
Scientific Reports (2022)
-
Childhood Trauma and Epigenetics: State of the Science and Future
Current Environmental Health Reports (2022)
-
Epigenetics as a Biomarker for Early-Life Environmental Exposure
Current Environmental Health Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
