
Abstract
Macrophages expressing C–C chemokine receptor type 2 (CCR2) infiltrate the central and peripheral neural tissues of amyotrophic lateral sclerosis (ALS) patients. To identify the functional role of CCR2+ macrophages in the pathomechanisms of ALS, we used an ALS animal model, mutant Cu/Zn superoxide dismutase 1G93A (mSOD1)-transgenic (Tg) mice. To clarify the CCR2 function in the model, we generated SOD1G93A/CCR2Red fluorescence protein (RFP)/Wild type (WT)/CX3CR1Green fluorescence protein (GFP)/WT-Tg mice, which heterozygously express CCR2-RFP and CX3CR1-GFP, and SOD1G93A/CCR2RFP/RFP-Tg mice, which lack CCR2 protein expression and present with a CCR2-deficient phenotype. In mSOD1-Tg mice, mSOD1 accumulated in the sciatic nerve earlier than in the spinal cord. Furthermore, spinal cords of SOD1G93A/CCR2RFP/WT/CX3CR1GFP/WT mice showed peripheral macrophage infiltration that emerged at the end-stage, whereas in peripheral nerves, macrophage infiltration started from the pre-symptomatic stage. Before disease onset, CCR2+ macrophages harboring mSOD1 infiltrated sciatic nerves earlier than the lumbar cord. CCR2-deficient mSOD1-Tg mice showed an earlier onset and axonal derangement in the sciatic nerve than CCR2-positive mSOD1-Tg mice. CCR2-deficient mSOD1-Tg mice showed an increase in deposited mSOD1 in the sciatic nerve compared with CCR2-positive mice. These findings suggest that CCR2+ and CX3CR1+ macrophages exert neuroprotective functions in mSOD1 ALS via mSOD1 clearance from the peripheral nerves.
Similar content being viewed by others


Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by the loss of upper and lower motor neurons. Although the majority of ALS cases are sporadic, approximately 10% are inherited1. Sporadic and familial ALS are clinically and pathologically similar. Approximately 20% of familial cases are linked to autosomal dominant mutations in the Cu/Zn superoxide dismutase 1 (SOD1) gene2. Hallmark pathological features in sporadic and familial ALS include the presence of axonal spheroids and perikaryal accumulation of inclusion bodies comprising neuronal intermediate filament proteins, such as neurofilaments and peripherin3,4.
Although the exact mechanism of ALS remains elusive, protein misfolding and aggregation have been implicated as contributing factors to motor neuron death5. This abnormal protein aggregation is thought to trigger non-cell autonomous neuronal cell death via glia-mediated mechanisms6. In ALS, activated microglia, macrophages, and astrocytes may be neuroprotective in the early stage but become pro-inflammatory and neurotoxic in the later stage when damage-associated molecular patterns are released from injured motoneurons with accumulated misfolded proteins, and they promote the pro-inflammatory activation of glial cells7,8. We and others have reported a variety of immune abnormalities in ALS, including increased cerebrospinal fluid (CSF) pro-inflammatory cytokines/chemokines, such as interleukin (IL)-1β, IL-12, IL-17, tumor necrosis factor-α, interferon-γ, C–C motif chemokine ligand (CCL) 2, CCL4, CCL11, C-X-C motif chemokine ligand (CXCL) 8, and CXCL109, increased serum IL-6, IL-17, and CCL210,11,12, and increased circulating IL-13-producing T cells13. However, it remains unclear whether these immune abnormalities cause disease or are a consequence of disease and whether they are neurotoxic or neuroprotective according to the disease stage.
Monocyte-macrophage lineage cells are heterogenous and express distinct chemokine receptors14. C–C chemokine receptor 2 (CCR2) is expressed by peripheral monocytes/macrophages, in addition to T cells, basophils, and immature dendritic cells15, whereas resident macrophages, such as microglia in the central nervous system (CNS), Kupffer cells in the liver, and Langerhans cells in the skin, express high levels of C-X3-C chemokine receptor 1 (CX3CR1)16. Notably, a CCL2/CCR2-dependent immunological pathway has been implicated in ALS. Patients had an increased level of CCL2 in the CSF and serum about 1 year before the onset of ALS9,17. Furthermore, spinal cord tissues from mutant SOD1 (mSOD1) transgenic ALS model mice also showed an increased level of CCL2 mRNA18,19. Conversely, CCR2 expression in the peripheral blood monocytes of ALS patients was significantly decreased compared with healthy controls20,21. These results collectively suggest the initial recruitment of peripheral monocytes/macrophages expressing CCR2 to neural tissues. However, it remains to be established whether these monocytes/macrophages exert protective or deleterious effects in ALS pathogenesis.
To address this issue in the present study, we aimed to clarify the functions of CCR2-bearing monocytes/macrophages recruited from the peripheral blood to the neural tissues in ALS. For this purpose, genetically labeled transgenic “Red-Green” SOD1G93A mice, which express CCR2-red fluorescence protein (RFP) and CX3CR1-green fluorescence protein (GFP) heterozygously, were used to characterize monocyte lineage cell dynamics. Furthermore, the role of CCR2 in ALS was explored using SOD1G93A/CCR2RFP/RFP mice, which possess a CCR2-deficient phenotype.
Results
mSOD1 accumulates in peripheral nerves earlier than in the spinal cord
mSOD1 immunostaining of SOD1G93A mice indicated the deposition of mSOD1 was absent in the spinal cord at 4 weeks of age and mainly present in the anterior horns at 12 weeks of age (onset stage) (Fig. 1a, b). mSOD1 protein was accumulated and spread along the pyramidal tracts and eventually extended into whole spinal cord cross-sectional areas at 20 weeks of age (moribund period). Conversely, the accumulation of mSOD1 in peripheral nervous system (PNS) tissues was detected in the spinal roots and dorsal root ganglia (DRG) as early as 4 weeks of age and was increased abundantly in the ventral roots compared with the dorsal roots. Double immunostaining for Iba1 and mSOD1 demonstrated Iba1+ foamy macrophages in the sciatic nerve were filled with mSOD1 protein at 20 weeks of age (Fig. 1c).

Immunostaining for mSOD1 and Iba1 in neural tissues. (a) Immunostaining for human mSOD1 in the lumbar spinal cord, dorsal root ganglia (DRG), ventral root (VR), and dorsal root (DR) from CCR2RFP/WT/CX3CR1GFP/WT (non-mSOD1 type) and SOD1G93A/CCR2RFP/WT/CX3CR1GFP/WT (mSOD1-Tg) mice. Accumulation of mSOD1 protein in the DRG, VR, and DR prior to accumulation in the lumbar cord was apparent at 4 weeks of age. In the spinal cord, mSOD1 protein started to accumulate along the intramedullary lower motoneuron axonal tracts (arrow) at 12 weeks of age. (b) Schemas depict mSOD1 protein accumulation over the disease course in the mSOD1-Tg ALS mouse model. (c) Double immunostaining for Iba1 (green) and mSOD1 (red) in the sciatic nerves of a 20-week-old mSOD1-Tg mouse. All Iba1+ macrophages (arrowhead) harbored mSOD1 protein. Scale bars: (a) 200 μm (lumbar spinal cord), and 100 μm (DRG, VR, and DR): (c) 10 μm.
Microglial responses commence in the spinal anterior horns at clinical disease onset
Microscopic analysis of CX3CR1 and CCR2 in the spinal cord from SOD1G93A/CCR2RFP/WT/CX3CR1GFP/WT mice revealed that the accumulation of CX3CR1+ microglia occurred mainly in the lumbar anterior horns at 12 weeks of age (at the time of clinical disease onset) without the infiltration of CCR2+ peripheral immunocytes (Fig. 2a). At 16 weeks of age (at the progressive stage), microglial activation became more robust, particularly along the intramedullary ventral roots, whereas CCR2+ cells were rarely observed. At 20 weeks of age (the moribund stage), CX3CR1+ activated microglia/macrophages without a foamy shape were present throughout the entire spinal cord section, whereas fewer CCR2+ cells had infiltrated the lesion (Fig. 2a).

CX3CR1 and CCR2 expression in the lumbar spinal cord and sciatic nerve. (a) Low magnification fluorescence images of CCR2RFP/WT/CX3CR1GFP/WT (non-mSOD1 type) and SOD1G93A/CCR2RFP/WT/CX3CR1GFP/WT (mSOD1-Tg) mouse spinal cord. At 12 weeks of age, CX3CR1+ (green) but not CCR2+ (red) cells were sparsely visible only in the spinal anterior horns (arrowhead). CX3CR1+ cells markedly increased as the disease progressed from 16 to 20 weeks of age, whereas CCR2+ peripheral immunocytes rarely infiltrated at 20 weeks of age. Infiltration of CX3CR1+ cells along the intramedullary lower motoneuron axonal tracts was prominent at 16 weeks of age (arrow). (b) Low magnification fluorescence images of CCR2RFP/WT/CX3CR1GFP/WT (non-mSOD1 type) and SOD1G93A/CCR2RFP/WT/CX3CR1GFP/WT (mSOD1-Tg) mouse sciatic nerves. In the sciatic nerves of a mSOD1-Tg mouse, CX3CR1+ cell infiltrations were visible as early as 4 weeks of age and CX3CR1+CCR2+ cells appeared at 8 weeks of age (pre-symptomatic stage). These infiltrated cells showed a foamy appearance (inset in the 8 weeks merged image). Such inflammatory infiltrates were not seen in non-mSOD1 type mice. (c) The quantitative analysis of CCR2+ CX3CR1− (red), CCR2−CX3CR1+ (green), and CX3CR1+CCR2+ (yellow) areas (%) in sciatic nerves. In SOD1G93A mice, numbers of all types of macrophages (CCR2−CX3CR1+ green, and CCR2+CX3CR1+ yellow macrophages) increased as the disease progressed, whereas CCR2+CX3CR1− red immune cells did not. Scale bars: (a) 500 μm; (b) 50 μm and 10 μm (inset). n.s. = not significant. *p < 0.05; **p < 0.001; ***p < 0.0001.
CX3CR1+ and CCR2+ cells infiltrate peripheral nerves earlier than in the spinal cord
Because a previous study reported the earlier deposition of mSOD1 protein in the lumbar spinal cord, sciatic nerves, and gastrocnemius muscles compared with the cervical spinal cord22, we immunohistochemically compared monocyte/macrophage infiltration into the sciatic nerves of SOD1G93A/CCR2RFP/WT/CX3CR1GFP/WT and CCR2RFP/WT/CX3CR1GFP/WT mice (non-mSOD1 type littermate). In the sciatic nerve of CCR2RFP/WT/CX3CR1GFP/WT mice, CX3CR1+ resident macrophages were evenly scattered, whereas CCR2+ immunocytes were rarely seen at 16 weeks of age (Fig. 2b). We did not observe CX3CR1+CCR2+ cells in the sciatic nerves of non-mSOD1 type mice. Conversely, the sciatic nerves of SOD1G93A/CCR2RFP/WT/CX3CR1GFP/WT mice showed a marked increase of CX3CR1+CCR2− (green) cells, but not CX3CR1−CCR2+ (red) or CX3CR1+CCR2+ (yellow) cells at 4 weeks of age. At 8 weeks of age (pre-onset stage), CX3CR1+CCR2+ (yellow) cells appeared in the sciatic nerve, and successively higher numbers appeared at 12 to 16 weeks of age (onset to progressive phases) (Fig. 2b, c). The increase ratio was more prominent for green cells compared with yellow cells. Many green cells had a foamy appearance (Fig. 2b inset).
CCR2 deficiency aggravates the clinical course of SOD1G93A ALS mice
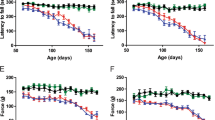
To clarify the effects of CCR2 deletion upon mSOD1-ALS, SOD1G93A/CCR2RFP/WT mice (CCR2-positive SOD1G93A mice, n = 24) and SOD1G93A/CCR2RFP/RFP mice (CCR2-deficient SOD1G93A mice, n = 18) were compared by measuring body weights, rotarod test, grip strength, and ALS-Therapy Development Institute (TDI) scores. CCR2-deficient SOD1G93A mice showed a 6-day acceleration in progression to the moribund stage (164.0 ± 1.48 days vs. 170.4 ± 2.06 days, p = 0.0047; Fig. 3a) compared with CCR2-positive SOD1G93A mice. Clinical signs were also significantly exacerbated in CCR2-deficient mice as determined by the rotarod test (p < 0.05; 12 weeks to 17 weeks; Fig. 3b), grip strength (p < 0.05; 13 weeks to 15 weeks; Fig. 3c), and ALS-TDI scores (area under the curve from 7 to 19 weeks of age: p = 0.031; Fig. 3d, e) compared with CCR2-positive SOD1G93A littermates. The peak body weight was reached earlier in CCR2-deficient SOD1G93A mice than in CCR2-positive SOD1G93A littermates, although the difference was not statistically significant (13.83 ± 0.34 weeks vs. 14.83 ± 0.35 weeks, respectively, p = 0.188; Fig. 3f).

Clinical manifestations of CCR2-positive and CCR2-deficient SOD1G93A mice. (a) Median survival time, (b) rotarod test, (c) grip strength, (d) ALS-TDI score, and (e) area under the curve (AUC) of ALS-TDI scores measured from 7 to 17 weeks of age in (d). (f) Mean age of disease onset was defined as when mice reached peak body weight (CCR2-positive SOD1G93A mice, n = 24, and CCR2-deficient SOD1G93A mice, n = 18). The log-rank test was used to compare median survival time and onset time by body weight. Student’s t test was used for statistical comparisons of rotarod test and grip strength. The Mann–Whitney U-test was used for statistical comparisons of the AUC of the ALS-TDI scores. *p < 0.05; **p < 0.001.
CCR2 deficiency accelerates mSOD1 accumulation in peripheral nerves
Immunohistochemical analysis with anti-human specific mSOD1 antibody revealed a significant increase in mSOD1 protein aggregation in the sciatic nerves of CCR2-deficient SOD1G93A mice compared with CCR2-positive littermates (p = 0.032; Fig. 4a, b) at 12 weeks of age (onset period). The accumulation of mSOD1 protein was also confirmed by western blotting (Supplementary Fig. 1). In the sciatic nerves, mSOD1 protein immunostaining was surrounded by the myelin sheath in CCR2-positive SOD1G93A mice. Conversely, CCR2-deficient SOD1G93A mice demonstrated immunostaining of mSOD1 protein aggregation inside and outside the myelin sheath (Fig. 4c), which suggests that excessive mSOD1 protein accumulation had overflowed into the myelin sheath (arrows in Fig. 4c). Conversely, there were no significant differences in mSOD1 protein aggregation in the lumbar spinal cord of CCR2-deficient SOD1G93A mice and CCR2-positive littermates (p = 0.77; Fig. 4d, e) at 12 weeks of age (onset period).

Aggravation of mSOD1 accumulation in the nerve by CCR2 ablation. (a) Immunostaining for mSOD1 in the sciatic nerves of CCR2-deficient SOD1G93A mice and CCR2-positive littermates. (b) Accumulation of mSOD1 protein was significantly increased in CCR2-deficient SOD1G93A mice than in CCR2-positive SOD1G93A mice at 12 weeks of age (p = 0.032, n = 5). (c) Double immunostaining for mSOD1 and Schwann cells indicated a greater accumulation of mSOD1 protein in the sciatic nerves of CCR2-deficient SOD1G93A mice showing an overflow of mSOD1 protein from Schwann cells and in the myelin sheath (arrows) compared with CCR2-positive SOD1G93A mice at 12 weeks of age. (d) Immunostaining for mSOD1 protein in the lumbar spinal cord of CCR2-deficient SOD1G93A mice and CCR2-positive littermates at 12 weeks of age. (e) There was no difference in the mSOD1+ area (%) between the two mouse genotypes at 12 weeks of age (p = 0.85, n = 6). The unpaired t test was used for statistical comparisons. Scale bars: (a) 20 μm; (c) 10 μm; (d) 100 μm. *p < 0.05. n.s. = not significant.
CCR2 deficiency worsens anterior horn cell loss and axonal deformation
The loss of anterior horn cells as evaluated by NeuN immunostaining was facilitated in CCR2-deficient SOD1G93A mice compared with CCR2-positive SOD1G93A mice (Fig. 5a). Significantly fewer anterior horn cell numbers were observed in CCR2-deficient SOD1G93A mice than in CCR2-positive littermates at 12 weeks of age (p = 0.033) (Fig. 5b). Moreover, SMI 31/32 immunostaining of peripheral nerve transverse sections revealed that the axons of CCR2-deficient SOD1G93A mice, but not CCR2-positive SOD1G93A mice, already showed crescent-like deformation at 8 weeks of age (pre-onset period) (Fig. 5c). The aspect ratio (calculated as the ratio between major and minor axis lengths) was significantly higher in CCR2-deficient SOD1G93A mice than in CCR2-positive SOD1G93A mice (aspect ratio: 1.779 vs. 1.683, p = 4.11 × 10−16; Fig. 5d), which indicates accelerated axonal deformation in CCR2-deficient SOD1G93A mouse peripheral nerves. Electron microscopy also confirmed that axons were more deranged in CCR2-deficient SOD1G93A mice compared with CCR2-positive SOD1G93A mice, whereas foamy macrophages filled with myelin and other cell debris were more frequently observed in CCR2-positive SOD1G93A mice compared with CCR2-deficient SOD1G93A mice (Fig. 6).

Facilitation of neuro-axonal degeneration by CCR2 ablation. (a) NeuN immunostaining revealed increased anterior horn neuronal cell loss in CCR2-deficient SOD1G93A mice compared with CCR2-positive SOD1G93A mice at 12 and 20 weeks of age. Inset areas in the low magnified images are enlarged under each figure. (b) CCR2-deficient SOD1G93A mice showed a significant decrease in NeuN+ cell numbers compared with CCR2-positive SOD1G93A mice at 12 weeks of age (p = 0.033, n = 12). (c) SMI 31/32 immunostaining of sciatic nerve transverse sections indicated axons in CCR2-deficient SOD1G93A mice had crescent-shaped deformation compared with CCR2-positive SOD1G93A littermates at 8 weeks of age. (d) The aspect ratio (the ratio of the major to the minor axis length) of CCR2-deficient SOD1G93A mouse axons was significantly larger than that of CCR2-positive SOD1G93A mouse axons at 8 weeks of age (p < 0.001, n = 5). Scale bars: (a) 100 μm and 50 μm; c, 20 μm. The horizontal line represents the mean value in (b, d). *p < 0.05; **p < 0.001.

Electron microscopic findings of the sciatic nerve. (a) The longitudinal section of the sciatic nerve from a CCR2-positive SOD1G93A mouse at 12 weeks of age shows preserved axonal structure and some derangement of the Schmidt-Lanterman incisures (arrow). (b) Cross-section of the sciatic nerve from a CCR2-positive SOD1G93A mouse at 20 weeks of age with relatively preserved axons and some vesiculation of the myelin sheath. (c) Foamy macrophages containing cell and myelin debris are visible adjacent to the axons in the longitudinal section of the sciatic nerve from a CCR2-positive SOD1G93A mouse at 12 weeks of age (arrowhead). (d) Longitudinal section of the sciatic nerve from a CCR2-deficient SOD1G93A mouse at 12 weeks of age with disruption of intra-axonal structures. (e) Cross-section of the sciatic nerve from a CCR2-deficient SOD1G93A mouse at 20 weeks of age with many disrupted, shrunken axons and marked vesiculation of the myelin sheath. Scale bar: 5 μm.
CCR2 deficiency diminishes anti-inflammatory M2 macrophage infiltration into the peripheral nerves
As shown in Fig. 7a, CCR2+ cell infiltration into the sciatic nerve was markedly diminished in CCR2-deficient SOD1G93A mice compared with CCR2-positive littermates at 12 (onset stage) and 20 weeks of age (moribund stage) (Fig. 7a). As a result, there were significantly fewer CCR2-RFP positive cells in the sciatic nerves of CCR2-deficient mice compared with CCR2-positive SOD1G93A mice at 12 weeks of age (p = 0.021; Fig. 7b). In addition, Iba1 immunostaining also indicated a significant decrease in Iba1+ macrophages in the sciatic nerves of CCR2-deficient SOD1G93A mice compared with CCR2-positive SOD1G93A mice at 12 weeks of age (p = 0.047; Fig. 7c, d). To further characterize the phenotype of infiltrating macrophages in the peripheral nerves of mSOD1 ALS mice, immunostaining for arginase-1 (Arg-1), a marker of anti-inflammatory M2 macrophages, and inducible nitric oxide synthase (iNOS), a marker of pro-inflammatory M1 macrophages, was performed. In the sciatic nerves of both mouse genotypes, infiltrated foamy macrophages were immunopositive for Arg-1 but negative for iNOS (Fig. 7e). CCR2 positive foamy-macrophages that were immunopositive for CD68 contained Arg-1 (Fig. 7f; Supplementary Fig. 2). Regarding T cell infiltration into the peripheral nerves, histological analysis indicated CD3+ T cells were rarely detected in the sciatic nerves of CCR2+ or CCR2− SOD1G93A mice and that there was no significant difference in cell numbers between these mouse strains (Supplementary Fig. 3).

Inhibition of macrophage infiltration into the sciatic nerve by CCR2 ablation. (a) Fluorescent microscopic analysis of unstained sciatic nerve tissues of CCR2-positive non-mSOD1 type mice, CCR2-positive SOD1G93A mice, and CCR2-deficient SOD1G93A mice. The influx of CCR2-RFP+ cells into the sciatic nerve was markedly diminished in CCR2-deficient SOD1G93A mice than in CCR2-positive littermates. (b) The number of CCR2-RFP+ cells was significantly reduced in CCR2-deficient SOD1G93A mice compared with CCR2-positive littermates (p = 0.021, n = 9). (c) Immunostaining for Iba1 in sciatic nerves shows a marked reduction of CCR2+ cell infiltrates in CCR2-deficient SOD1G93A mice compared with CCR2-positive littermates. (d) The number of Iba-1+ cells was significantly reduced in CCR2-deficient SOD1G93A mice compared with CCR2-positive littermates (p = 0.047, n = 6). (e) Immunostaining for arginase-1 (Arg-1), an M2 marker, and inducible nitric oxide synthase (iNOS), an M1 marker, of the sciatic nerve, demonstrated infiltrated macrophages were Arg-1+ but iNOS-, which suggests an M2-deviated phenotype. Inset in (e) indicates the foamy appearance of Arg-1+ cells. (f) Foamy macrophages with Arg-1 are CCR2-positive. Scale bars: (a) 100 μm and 20 μm (inset); (c) 50 μm; (e) 50 μm; (f) 20 μm. The horizontal line in (b, d) represents the mean value. The unpaired t test was used for statistical comparisons. *p < 0.05.
Differentiated macrophages are decreased in the sciatic nerves of CCR2-deficient mice
Flow cytometric analysis of cells isolated from sciatic nerves revealed comparable leukocyte ratios between CCR2+ and CCR2− mSOD1-Tg mice, except for a decrease in CD11b+/CD11c+ macrophages in CCR2-deficient mice (CD11b+/CD11c+ cell population ratio in CD45+ gate (%) (CCR2+mSOD1 vs. CCR2-mSOD1, mean ± SEM = 25.40 ± 2.577 vs. 12.67 ± 1.619, p = 0.0111). There were no significant differences in the CD45+CD3+ T cell population ratio in the sciatic nerves of CCR2+ and CCR2− mSOD1-Tg mice (Supplementary Fig. 4).
CCL2 production by Schwann cells is unchanged by CCR2 deficiency
Double immunostaining with anti-Schwann cell antibody (human peripheral nerve extract antigen, mouse IgM antibody)23 and anti-CCL2 antibody demonstrated that CCL2 was mainly co-localized with Schwann cells in the sciatic nerves of SOD1G93A mice at 16 weeks, regardless of the presence or absence of CCR2 (Fig. 8a). This indicated that Schwann cells are the primary source of CCL2 in peripheral nerves. The numbers of CCL2+ cells in the sciatic nerve were not significantly different between CCR2-deficient and CCR2-positive SOD1G93A mice (p = 0.92; Fig. 8b). The anti-Schwann cell antibody we used in this study was a mouse IgM antibody that uses human peripheral nerve extract as its antigen.

CCL2 production by Schwann cells. (a) Double immunostaining of CCR2-deficient and CCR2-positive SOD1G93A mouse sciatic nerves at 16 weeks with anti-Schwann cell and anti-CCL2 antibodies. The merged images of both antibodies indicate the production of CCL2 by Schwann cells in both mouse genotypes. Scale bar: 20 μm. (b) The CCL2+ area (%) was not significantly different between CCR2-deficient and CCR2-positive SOD1G93A mice. The horizontal line represents the mean value. The unpaired t test was used for statistical comparisons (p = 0.92, n = 9). n.s. = not significant.
Phagocytic activity of isolated macrophages is unaltered by CCR2 deficiency
Finally, we measured the phagocytic activity of macrophages isolated from CCR2RFP/RFP SOD1G93A mice, CCR2RFP/WT SOD1G93A mice, CCR2RFP/RFP mice, and CCR2RFP/WT mice. There was no difference in phagocytic activity among these mice in vitro (Supplementary Fig. 5), which suggests that a deficiency of CCR2 or the presence of mSOD1 does not influence the phagocytic activity of isolated macrophages.
Discussion
The role of peripheral blood-borne macrophages bearing CCR2 recruited into the neural tissues has long been a critical question in ALS. To address this issue, we studied mSOD1 ALS mice with genetically labeled macrophages or with CCR2 deficiency. We found that before the onset of clinical symptoms, CCR2+ macrophages that phagocytized mSOD1 protein had already infiltrated the peripheral nerves much earlier than into the spinal cord. Furthermore, CCR2 ablation clinically accelerated the disease progression and worsened the pathology, as determined by NeuN+ neuronal loss in the spinal anterior horns and axonal derangement in the peripheral nerves. CCR2 ablation also markedly increased the accumulation of mSOD1 protein in the peripheral nerves and, to a lesser extent, in the spinal cord compared with CCR2-positive mice. Flow cytometric analysis revealed a comparable number of CD3+ T cells were detected in the sciatic nerves of 8-week-old mSOD1-Tg mice with and without CCR2. However, we also detected the suppressed infiltration of the CD11b+/CD11c+ cell population, which indicates that activated macrophages derived from monocytes24 were decreased in the sciatic nerves of CCR2-deficient mice. A decreased infiltration of CCR2+ macrophages, which phagocytized mSOD1 protein and expressed Arg-1, an M2 marker, but not iNOS, an M1 marker, in the peripheral nerves was also observed. These findings suggest that CCR2+ macrophages recruited into the peripheral nerves from the blood exert neuroprotective functions on the lower motor neurons in mSOD1 ALS and that the clearance of abnormal mSOD1 protein from peripheral nerves by these cells is a hitherto underestimated host protective mechanism (Supplementary Fig. 6).
The SOD1G93A/CCR2RFP/WT/CX3CR1GFP/WT mice started to deteriorate at 12 weeks of age and died at 21 weeks of age, which is similar to the reported clinical course of mSOD1G93A ALS mice22. In the SOD1G93A/CCR2RFP/WT/CX3CR1GFP/WT mice, we observed that mSOD1 accumulated in the peripheral nerves much earlier (4 weeks of age) than in the spinal cord (12 weeks of age), which is in accord with a previous report describing the earlier accumulation of mSOD1 protein in the sciatic nerve compared with the lumbar spinal cord (2.6-fold increase in the sciatic nerve vs. 1.8-fold increase in the lumbar cord at 30 days of age compared with at birth, and remaining consistently higher in the sciatic nerve than in the lumbar cord until 120 days of age)22. These observations support distal axonopathy as a primary mechanism of the lower motor neuron death in mSOD1 ALS6,25. We also confirmed the accumulation of mSOD1 in the DRG and, to a lesser extent, in the dorsal roots as previously reported, which explains the sensory system involvement observed in mSOD1 ALS patients and model animals26. Because DRGs are bipolar cells, we hypothesized that mutant protein transported by an afferent mechanism is not cleared in CNS regions where there is no or little peripheral macrophage infiltration, unlike the sciatic nerve, which harbors high numbers of peripheral macrophages. It was suggested that there may be differences in the characteristics of axonal transport between motor and sensory neurons in the SOD1 mouse model related to differences in dynein and dynactin functions27.
In line with the earlier accumulation of mSOD1 protein in the peripheral nerves, we found increased CX3CR1+ macrophages infiltration into the sciatic nerve as early as 4 weeks of age and the subsequent infiltration of CX3CR1+CCR2+ macrophages at 8 weeks of age. CX3CR1+ macrophages, which were observed at 4 weeks of age, are considered resident macrophages, whereas CX3CR1+CCR2+ macrophages are thought to be peripheral blood-borne macrophages. CCR2 single positive cells are considered peripheral blood-derived immune cells, other than monocytes/macrophages. We also found a rapid increase in CX3CR1+CCR2− green cell numbers in peripheral nerves in the progressive phase of the disease. Also, the appearance of green cells was different between 4 weeks (thin shape) and 8 weeks (foamy shape). Figure 2c shows an increase of CX3CR1+CCR2+ yellow cells. RFP expression was reduced after monocytes infiltrated tissues because its half-life is up to 4.6 days28. As shown in Supplementary Fig. 7, peripheral yellow monocytes infiltrate the sciatic nerve and then differentiate into macrophages, which lose their red color, which results in green foamy cells. The increase of yellow cells at 12 and 16 weeks indicates the acceleration of monocyte infiltration into lesions. We assume that the green foamy cells were initially infiltrated as blood-borne monocytes with yellow color, but soon after the CCR2 expression was down-regulated along with RFP, which resulted in green foamy cells. These findings are in line with previous reports that CCR2 on activated monocytes is rapidly internalized, and that the synthesis of CCR2 is down-regulated15. Our results are compatible with previous results showing CD68+ macrophages infiltrated the ventral roots of mSOD1G93A mice at 60 days of age22. Although it was unclear whether these cells were protective or worsened disease progression, we hypothesize these macrophages scavenge mSOD1 protein to protect peripheral nerves. This notion is supported by the observations that these macrophages were full of mSOD1 protein and that a deficiency of these cells by CCR2 ablation markedly facilitated mSOD1 accumulation in the nerves and promoted the death of the lower motor neurons. Flow cytometric analysis revealed a decrease of the CD11b+/CD11c+ cell population in CCR2 deficient mice. Because CCL2 induces the chemotaxis of CCR2 positive cells and induces the differentiation of monocytes, this disparity in the CD11b+/CD11c+ cell ratio might result from the lack of CCL2 signaling in CCR2 deficient monocytes. Activated macrophages are subcategorized into several phenotypes, including M1 pro-inflammatory and M2 anti-inflammatory. The direction of activation induced by CCL2 depends on the inflammatory circumstances29,30. We confirmed that the foamy macrophages were all Arg-1 positive M2 phenotype, which indicated that the lack of CCL2 signaling in CCR2RFP/RFP SOD1G93A mice decreased the infiltration of CCR2-positive cells into the sciatic nerve and inhibited macrophage differentiation into M2 macrophages.
We confirmed that CCL2 production by Schwann cells, which are the primary source of CCL2 in nerves31,32, was unaffected in CCR2RFP/RFP SOD1G93A mice. Thus, our findings are in accord with previous reports showing that CCL2 expression in the sciatic nerve was not altered by CCR2 gene deletion33 and that in peripheral nerve injury, CCR2-deficient mice had decreased macrophage accumulation in the sciatic nerve and DRG34,35. Whether CCR2 deficiency reduces phagocytic capacity is controversial36,37; however, we confirmed that the phagocytic activity of the isolated macrophages was unaffected by CCR2 ablation, at least in SOD1G93A/CCR2RFP/RFP mice. Collectively, these findings indicate that the impaired migratory activity and the inhibition of M2-directed differentiation of macrophages related to the lack of CCL2-CCR2 signaling in the inflammatory milieu where abnormal proteins are accumulated contributed to the insufficient clearance of misfolded mSOD1 protein in peripheral nerves, which results in the accelerated loss of anterior horn cells and exacerbated disease in CCR2-deficient SOD1G93A mice. In other neurodegenerative diseases, CCR2 deficiency was also reported to accelerate disease38,39. Notably, CCR2 deficiency particularly aggravated amyloid β clearance in an Alzheimer’s disease animal model, thereby inducing accelerated deterioration38,39. Taken together, these findings suggest CCR2+ peripheral blood-borne macrophages clear abnormal proteins, at least in the early stage of the disease, contributing to disease protection in neurodegenerative disorders, such as ALS, caused by abnormal protein accumulation.
CCR2 is abundantly expressed in M1 pro-inflammatory macrophages. Thus, CCR2 ablation is likely to inhibit M1 macrophage mobilization from peripheral blood to nerves. Given the pro-inflammatory properties of M1 macrophages, the neuroprotective action of peripheral blood-borne CCR2+ M1 macrophages in mSOD1 ALS is unexpected. However, importantly, these CCR2+ macrophages co-expressed CX3CR1 in the peripheral nerves of mSOD1 ALS mice, which suggests an intermediate phenotype, transitioning from pro-inflammatory M1 to anti-inflammatory M2 macrophages after invasion into the peripheral nerve. Macrophages were reported to switch phenotypes according to their microenvironment7,40. In peripheral nerve and spinal cord injury models, a transition from M1 to M2 phenotype was reported, and such intermediate macrophages had anti-inflammatory and neuroprotective properties41. Therefore, CCR2+ peripheral blood-borne macrophages that phagocytose mSOD1 may also be neuroprotective for peripheral nerves, thereby extending lower motor neuron survival time in mSOD1 ALS mice. There are several possibilities why the pathologic process still progresses even in the presence of macrophage clearance of misfolded mSOD1. Upper motor neurons are behind the blood–brain barrier, which may disturb macrophage clearance mechanisms in the early stage. Our results showed that CCR2+ cell infiltration into the spinal cord was only observed in the end-stage. Furthermore, there may be other neuropathic pathways in which the macrophage clearance mechanism does not work. The clinical course of CCR2+ mSOD1 mice eventually catches up at later periods. We hypothesized that this was related to the accumulation of mSOD1 protein in upper motor neurons because macrophage infiltration is blocked by the blood–brain barrier, and thus mSOD1 protein in the upper motor neuron cannot be removed.
Our study had several limitations. First, because of the small amount of CCR2-single positive red cells (T, B, and dendritic cells) in the sciatic nerve, we did not examine CCR2+ leukocyte involvement. It was reported that SOD1G93APU.−/− mice lacking CD3+ T cells had shorter life spans after transplantation with CCR2-deficient bone marrow compared with wild-type bone marrow transplantation42. CCR2+ T cells were thought to facilitate glial neuroprotection in this model. In our model, a detailed time-course study of T cell infiltration into the spinal cord and peripheral nerves is required in the future. Second, the characterization of infiltrated CCR2+ macrophages and their transition to the M2 phenotype was performed by immunohistochemistry because relatively small numbers of macrophages were present in the nerves. Future studies should characterize macrophages isolated from peripheral nerves by microarray or single-cell RNA sequencing.
In summary, our study revealed an under-recognized mechanism of abnormal protein clearance by CCR2+ macrophages from the peripheral nerves of mSOD1 ALS mice, which is beneficial to the host. Because lower motor neuron axons are present in peripheral nerves, which are more accessible to peripheral blood macrophages than CNS tissues that are tightly surrounded by the blood–brain barrier, peripheral nerves might be a novel therapeutic target for the cell therapy of ALS by removing abnormal proteins and delivering neuroprotective factors.
Materials and methods
Mice and ethical statement
Transgenic mice for the human SOD1G93A gene [B6SJL-TgN (SOD1*G93A) 1Gur/J; Stock Number: 002297] were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). They were crossed with C57BL/6J mice (Clea Japan, Tokyo, Japan) to maintain the strains, and hemizygous animals were used for the experiments. Transgenic mice harboring the human SOD1G93A gene were backcrossed to C57BL/6J mice for more than 15 generations. CCR2RFP mice [B6.129 (Cg)-Ccr2tm2.1Ifc/J; Stock Number: 017586], and CX3CR1GFP mice [B6.129P (Cg)-Ptprca Cx3cr1tm1Litt/LittJ; Stock Number: 008451] were also purchased from the Jackson Laboratory. Heterozygous SOD1G93A mice and CCR2RFP/RFP mice were crossed to obtain SOD1G93A/CCR2RFP/WT mice, and then SOD1G93A/CCR2RFP/WT mice and CCR2RFP/RFP mice were crossed to obtain SOD1G93A/CCR2RFP/WT mice and SOD1G93A/CCR2RFP/RFP mice. In CCR2RFP/RFP homozygotes, CCR2 alleles are inactivated (CCR2-deficient phenotype) whereas CCR2-RFP labeling is preserved. In addition, to create SOD1G93A/CCR2RFP/WT/CX3CR1GFP/WT mice, SOD1G93A/CCR2RFP/WT mice and CX3CR1GFP/GFP mice were crossed to obtain SOD1G93A/CCR2RFP/WT/CX3CR1GFP/WT mice. This dual heterozygous mouse expressing both reporter proteins and their receptors at functional levels has been widely used to differentiate resident microglia from blood-derived monocytes/macrophages in various neurodegenerative models15,43. Non-SOD1G93A-transgenic littermates were used as a non-ALS model phenotype. All animals were maintained in an air-conditioned, specific-pathogen-free room with a time-controlled lighting system. The handling and sacrifice of all animals were conducted according to the guidelines for the proper conduct of animal experiments published by the Science Council of Japan and the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines for animal research. Ethical approval for the study was granted by the Animal Care and Use Committee of Kyushu University (#A30-051).
Behavioral study
Body weights, performances in the rotarod test (ENV-576M; Neuroscience, Tokyo, Japan), grip strength (MK-380M, Muromachi-Kikai, Tokyo, Japan), and ALS-TDI neurological scoring44 were assessed once a week. For the assessment of motor function by rotarod, mice were habituated to stay on the stationary drum for 3 min before the training sessions. Habituation was repeated each time for 1 min just before the session. Mice were examined on the rotarod with an accelerating speed of 5 to 30 rpm over 300 s. The trials were performed three times, and the longest time was recorded. The time limit of each observation was 300 s. The time of disease onset was retrospectively determined as the time when the mice reached their peak body weight. For the assessment of grip strength, mice were lifted by the base of the tail and placed so that their front paws gripped the trapeze with their body horizontal. Each mouse was tested five times to obtain the best grip strength performance. The ALS-TDI neurological score was measured as follows: 0, normal gait is observed; 1, the hindlimb collapsed towards the lateral midline or trembled; 2, while walking, any part of the foot dragged along the cage bottom/table; 3, the hindlimb was not used for forwarding motion but was able to right itself within 10 s; and 4, rigid paralysis in the hindlimb and absence of righting reflex.
Immunohistochemistry
All animals were deeply anesthetized with sevoflurane and perfused intracardially with saline followed by cold 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). The lumbar cord, sciatic nerve, and ventral roots were subsequently removed, immersed for over 12 h in the same 4% PFA fixative at 4 °C, and processed for making paraffin-embedded materials or optimal cutting temperature compound-embedded frozen materials. Multiple 5-μm-thick paraffin-embedded sections and 10-μm-thick frozen sections were used for immunohistochemical staining. Paraffin-embedded sections were deparaffinized, and frozen sections were air-dried. Endogenous peroxidase activity was blocked by 3% H2O2 in methanol/PBS (1:1) for 10 min at room temperature. Sections were then incubated with primary antibodies at 4 °C overnight45. After rinsing, sections were subjected to either a streptavidin–biotin complex or an enhanced indirect immunoperoxidase method using Envision (Dako Cytomation, CA, USA). Immunoreactivity was detected using 3,3′-diaminobenzidine as the chromogen. The primary antibodies for mouse tissues are listed in Supplementary Table 1. For immunohistochemical staining, the sections were incubated with secondary antibodies conjugated to Alexa Fluor 488 or 594 (1:1000; Thermo Fisher, Rockford, IL, USA) and 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, Tokyo, Japan) to stain cell nuclei, and mounted with Permafluor (#TA-030-FM; Thermo Scientific, Fremont, CA, USA). Tissues were observed with a fluorescence microscope (BZ-X700, Keyence, Tokyo, Japan).
Western blotting
Sciatic nerves of mice were homogenized in 50 μl of lysis buffer containing 8% sodium dodecyl sulfate (SDS) and RIPA buffer (#16488-34; Nacalai Tesque, Kyoto, Japan). After centrifugation at 14,000×g for 10 min, the supernatants were collected. The protein concentration in the supernatant was measured using a DC protein assay kit (Bio-Rad, Tokyo, Japan). Total protein (6 μl each) was separated by SDS polyacrylamide gel electrophoresis (15%). Western blotting analysis was performed using anti-mSOD1 (500 ng/ml) according to a previously described method46. The density of each band was quantified using ImageJ version 1.8.0_112 (Windows version of NIH Image; downloaded from https://imagej.nih.gov/ij/download.html).
Image acquirement and quantification analysis
Immunofluorescence was captured by a fluorescence microscope (BZ-X700). Quantification of immunofluorescence was performed using ImageJ version 1.8.0_112 using at least five lumbar spinal cord sections or five peripheral nerve sections for each animal in each group using the area fraction technique as previously described47.
Electron microscopy
The animals were perfused intracardially with saline followed by cold 4% paraformaldehyde. The sciatic nerve was prefixed with a fixation buffer (2.5% glutaraldehyde, 0.1 M sucrose, 3 mM CaCl2, and 0.1 M sodium cacodylate, pH 7.4) overnight at 4 °C. After being rinsed in PBS, the tissue was post-fixed with 1% osmium tetroxide for 2 h, dehydrated in ethanol and propylene oxide, and embedded in Epon resin (Epon 812 resin kit; TAAB Laboratories, Aldermaston, Berkshire, UK). Ultrathin Sects. (80 nm) were stained with uranyl acetate for 5 min and with lead acetate for 10 min and then examined with a transmission electron microscope (Tecnai 20; FEI Company Japan Ltd, Tokyo, Japan)45.
Mononuclear cell isolation from the sciatic nerve and flow cytometric analysis
After the transcardial perfusion by ice-cold PBS, sciatic nerves were removed, minced with scissors, then suspended in RPMI media. Then, the minced pieces were further dissociated using a 1000-µl pipette. The cell suspension was passed into the FACS buffer through a 100-µm cell strainer. This step was repeated several times. The cell suspension was centrifuged at 800 × g at 4 °C for 5 min, the supernatant discarded, and the cell pellet resuspended in 1 ml FACS buffer48. For surface marker staining, cells were incubated with fluorochrome-conjugated antibodies against CD45, CD3, I-A/I-E, CD11b, and CD11c for 30 min at 4 °C and analyzed in a BD FACSVerse™ flow cytometer (Becton, Dickinson and Company, NJ). The percentage of CD45+ leukocytes, CD3+ T cells, I-A/I-E+ monocytes, CD11b+CD11c− macrophages, and CD11b+CD11c+ phagocytes was measured.
Cell culture
To analyze the phagocytic activity of monocytes in vitro, peripheral heparinized whole blood was isolated from mice. Then, 100 µl of whole blood was incubated with 10 µl of pHrodo Green E. coli BioParticles Conjugate (10 mg/ml; Thermo Fisher Scientific, MA) and 40 µl of Gibco RPMI 1640 medium (Thermo Fisher Scientific) at 37 °C and 5% CO2 for 30 min. After a washing step, the cells were analyzed by flow cytometry using a Sony SH-800 (Sony Corporation, Tokyo, Japan).
Statistical analysis
Data are expressed as the mean ± standard error of the mean (SEM). Pairwise comparisons between two groups were performed using the unpaired t test and log-rank test. Comparisons between the three groups used in Fig. 2c were performed by two-way repeated-measures analysis of variance (ANOVA) followed by Bonferroni posttests. Multiple comparisons in Suppl. Figure 2 were performed by one-way factorial ANOVA. Survival time was compared using the Kaplan–Meier method and the log-rank test. A value of p < 0.05 was considered statistically significant. Quantitative analysis for FCM in Suppl. Figure 4b were performed by unpaired t test. All statistical analyses were conducted using JMP pro 12 software (SAS Institute, Cary, NC). Graphical images were built using PRISM 9 software (GraphPad Software, CA).
Ethical approval
The handling and sacrifice of all animals were conducted according to the guidelines for the proper conduct of animal experiments published by the Science Council of Japan, as well as the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines for animal research. Ethical approval for the study was granted by the Animal Care and Use Committee of Kyushu University (#A30-051).
References
Turner, M. R. et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 12, 310–322 (2013).
Rosen, D. R. et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 (1993).
Tu, P. H. et al. Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc. Natl. Acad. Sci. USA 93, 3155–3160 (1996).
Xiao, S. & Jesse McLean, J. R. Neuronal intermediate filaments and ALS: a new look at an old question. Biochim. Biophys. Acta Mol. Basis Dis. 1762, 1001–1012 (2006).
Blokhuis, A. M., Groen, E. J. N., Koppers, M., Van Den Berg, L. H. & Pasterkamp, R. J. Protein aggregation in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 777–794 (2013).
Ilieva, H., Polymenidou, M. & Cleveland, D. W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 187, 761–772 (2009).
Beers, D. R. & Appel, S. H. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol. 18, 211–220 (2019).
Nardo, G. et al. New insights on the mechanisms of disease course variability in ALS from mutant SOD1 mouse models. Brain Pathol. 26, 237–247 (2016).
Tateishi, T. et al. CSF chemokine alterations related to the clinical course of amyotrophic lateral sclerosis. J. Neuroimmunol. 222, 76–81 (2010).
Ono, S., Hu, J., Shimizu, N., Imai, T. & Nakagawa, H. Increased interleukin-6 of skin and serum in amyotrophic lateral sclerosis. J. Neurol. Sci. 187, 27–34 (2001).
Fiala, M. et al. IL-17A is increased in the serum and in spinal cord CD8 and mast cells of ALS patients. J. Neuroinflammation 7, 1–14 (2010).
Baron, P. et al. Production of monocyte chemoattractant protein-1 in amyotrophic lateral sclerosis. Muscle Nerve 32, 541–544 (2005).
Shi, N. et al. Increased IL-13-producing T cells in ALS: Positive correlations with disease severity and progression rate. J. Neuroimmunol. 182, 232–235 (2007).
Dal-Secco, D. et al. A dynamic spectrum of monocytes arising from the in situ reprogramming of CCR2+ monocytes at a site of sterile injury. J. Exp. Med. 212, 447–456 (2015).
Yamasaki, R., Liu, L., Lin, J. & Ransohoff, R. M. Role of CCR2 in immunobiology and neurobiology. Clin. Exp. Neuroimmunol. 3, 16–29 (2012).
Italiani, P. & Boraschi, D. From monocytes to M1/M2 macrophages: phenotypical vs. functional differentiation. Front. Immunol. 5, 1–22 (2014).
Gupta, P. K., Prabhakar, S., Sharma, S. & Anand, A. A predictive model for amyotrophic lateral sclerosis (ALS) diagnosis. J. Neurol. Sci. 312, 68–72 (2012).
Beers, D. R. et al. Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain. Behav. Immun. 25, 1025–1035 (2011).
Kawaguchi-Niida, M., Yamamoto, T., Kato, Y., Inose, Y. & Shibata, N. MCP-1/CCR2 signaling-mediated astrocytosis is accelerated in a transgenic mouse model of SOD1-mutated familial ALS. Acta Neuropathol. Commun. 1, 1–12 (2013).
Mantovani, S. et al. Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process. J. Neuroimmunol. 210, 73–79 (2009).
Zhang, R. et al. MCP-1 chemokine receptor CCR2 is decreased on circulating monocytes in sporadic amyotrophic lateral sclerosis (sALS). J. Neuroimmunol. 179, 87–93 (2006).
Turner, B. J., Lopes, E. C. & Cheema, S. S. Neuromuscular accumulation of mutant superoxide dismutase 1 aggregates in a transgenic mouse model of familial amyotrophic lateral sclerosis. Neurosci. Lett. 350, 132–136 (2003).
Arai, H., Hirato, J. & Nakazato, Y. A novel marker of Schwann cells and myelin of the peripheral nervous system. Pathol. Int. 48, 206–214 (1998).
Rogers, P. B., Driessnack, M. G. & Hiltbold Schwartz, E. Analysis of the developmental stages, kinetics, and phenotypes exhibited by myeloid cells driven by GM-CSF in vitro. PLoS ONE 12, 1–18 (2017).
Fischer, L. R. et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240 (2004).
Tao, Q. Q., Wei, Q. & Wu, Z. Y. Sensory nerve disturbance in amyotrophic lateral sclerosis. Life Sci. 203, 242–245 (2018).
Ström, A. L. et al. Retrograde axonal transport and motor neuron disease. J. Neurochem. 106, 495–505 (2008).
Yamasaki, R. Distinct roles of microglia and monocytes in central nervous system inflammation and degeneration. Clin. Exp. Neuroimmunol. 5, 41–48 (2014).
Gschwandtner, M., Derler, R. & Midwood, K. S. More than just attractive: how CCL2 influences myeloid cell behavior beyond chemotaxis. Front. Immunol. 10, 1–29 (2019).
Roca, H., Varcos, Z. S., Sud, S., Craig, M. J. & Pienta, K. J. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J. Biol. Chem. 284, 34342–34354 (2009).
Bakst, R. L. et al. Inflammatory monocytes promote perineural invasion via CCL2-mediated recruitment and cathepsin B expression. Cancer Res. 77, 6400–6414 (2017).
Tofaris, G. K., Patterson, P. H., Jessen, K. R. & Mirsky, R. Denervated Schwann cells attract macrophages by secretion of leukemia inhibitory factor (LIF) and monocyte chemoattractant protein-1 in a process regulated by interleukin-6 and LIF. J. Neurosci. 22, 6696–6703 (2002).
Yuan, F. et al. CCR2 Gene deletion and pharmacologic blockade ameliorate a severe murine experimental autoimmune neuritis model of Guillain-Barré syndrome. PLoS ONE 9, e90463 (2014).
Lindborg, J. A., Mack, M. & Zigmond, R. E. Neutrophils are critical for myelin removal in a peripheral nerve injury model of wallerian degeneration. J. Neurosci. 37, 10258–10277 (2017).
Siebert, H., Sachse, A., Kuziel, W. A., Maeda, N. & Brück, W. The chemokine receptor CCR2 is involved in macrophage recruitment to the injured peripheral nervous system. J. Neuroimmunol. 110, 177–185 (2000).
Chen, M., Forrester, J. V. & Xu, H. Dysregulation in retinal para-inflammation and age-related retinal degeneration in CCL2 or CCR2 deficient mice. PLoS ONE 6, e22818 (2011).
Suresh, M. V. et al. Role of macrophage chemoattractant protein-1 in acute inflammation after lung contusion. Am. J. Respir. Cell Mol. Biol. 46, 797–806 (2012).
El Khoury, J. et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat. Med. 13, 432–438 (2007).
Naert, G. & Rivest, S. CC chemokine receptor 2 deficiency aggravates cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 31, 6208–6220 (2011).
Malyshev, I. & Malyshev, Y. Current concept and update of the macrophage plasticity concept: Intracellular mechanisms of reprogramming and M3 macrophage ‘switch’ phenotype. Biomed Res. Int. 2015, 341308 (2015).
Liu, P. et al. Role of macrophages in peripheral nerve injury and repair. Neural Regen. Res. 14, 1335–1342 (2019).
Beers, D. R., Henkel, J. S., Zhao, W., Wang, J. & Appel, S. H. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc. Natl. Acad. Sci. USA 105, 15558–15563 (2008).
Saederup, N. et al. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS ONE 5, e13693 (2010).
Hatzipetros, T. et al. A quick phenotypic neurological scoring system for evaluating disease progression in the SOD1-G93A mouse model of ALS. J. Vis. Exp. 2015, e53257 (2015).
Kobayakawa, Y. et al. Galectin-1 deficiency improves axonal swelling of motor neurones in SOD1G93A transgenic mice. Neuropathol. Appl. Neurobiol. 41, 227–244 (2015).
Fujisawa, T. et al. A novel monoclonal antibody reveals a conformational alteration shared by amyotrophic lateral sclerosis-linked SOD1 mutants. Ann. Neurol. 72, 739–749 (2012).
Evonuk, K. S. et al. Inhibition of system Xc—transporter attenuates autoimmune inflammatory demyelination. J. Immunol. 195, 450–463 (2015).
Hidmark, A. S., Nawroth, P. P. & Fleming, T. Analysis of immune cells in single sciatic nerves and dorsal root ganglion from a single mouse using flow cytometry. J. Vis. Exp. 2017, 1–8 (2017).
Acknowledgements
We appreciate assistance from The Research Support Center, Research Center for Human Disease Modeling, and Kyushu University Graduate School of Medical Sciences. We thank Dr. Mitsuru Watanabe and Ms. Eriko Matsuo from the Department of Neurology, Kyushu University, for the technical assistance in the flow cytometric analysis. We thank Ms. Sachiko Koyama and Hideko Noguchi from the Department of Neuropathology, Kyushu University, for excellent technical assistance in the histological analysis. We thank Mr. Tetsuo Kishi from the Department of Medicine, Kyushu University School of Medicine for the immunohistochemical analysis. We thank J. Ludovic Croxford, PhD, from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript.
Funding
This study was supported in part by JSPS KAKENHI Grants-in-Aid for Scientific Research (C) (Grant Number JP16K09694 and JP19K07963) from the Japan Society for the Promotion of Science.
Author information
Authors and Affiliations
Contributions
W.S., R.Y., Y.H., S.K., and J.K. conceived the experiments. All authors contributed to the experimental design. W.S., R.Y., Y.H., and S.K. performed the experiments and analyzed the results. R.Y. provided technical advice for the experiments. W.S., R.Y., Y.H., Y.K., N.I., T.M., and J.K. were involved in interpreting the results. W.S., R.Y., and J.K. drafted the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
R.Y. has received honoraria from Teijin Pharma, Ono Pharmaceutical, Takeda Pharmaceutical, Eisai, Novartis, Nihon Pharmaceutical, and CSL Behring; J.K. is a consultant for Biogen Japan and Medical Review, and has received honoraria from Bayer Healthcare, Mitsubishi Tanabe Pharma, Nobelpharma, Otsuka Pharmaceutical, Sanofi KK, Chugai Pharmaceutical Co. Ltd., Teijin Pharma, Novartis Pharma, and Medical Review; the remaining authors declare no conflicts of interest.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shiraishi, W., Yamasaki, R., Hashimoto, Y. et al. Clearance of peripheral nerve misfolded mutant protein by infiltrated macrophages correlates with motor neuron disease progression. Sci Rep 11, 16438 (2021). https://doi.org/10.1038/s41598-021-96064-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-96064-6
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
