
Abstract
Multi-locus genetic data are pivotal in phylogenetics. Today, high-throughput sequencing (HTS) allows scientists to generate an unprecedented amount of such data from any organism. However, HTS is resource intense and may not be accessible to wide parts of the scientific community. In phylogeography, the use of HTS has concentrated on a few taxonomic groups, and the amount of data used to resolve a phylogeographic pattern often seems arbitrary. We explore the performance of two genetic marker sampling strategies and the effect of marker quantity in a comparative phylogeographic framework focusing on six species (arthropods and plants). The same analyses were applied to data inferred from amplified fragment length polymorphism fingerprinting (AFLP), a cheap, non-HTS based technique that is able to straightforwardly produce several hundred markers, and from restriction site associated DNA sequencing (RADseq), a more expensive, HTS-based technique that produces thousands of single nucleotide polymorphisms. We show that in four of six study species, AFLP leads to results comparable with those of RADseq. While we do not aim to contest the advantages of HTS techniques, we also show that AFLP is a robust technique to delimit evolutionary entities in both plants and animals. The demonstrated similarity of results from the two techniques also strengthens biological conclusions that were based on AFLP data in the past, an important finding given the wide utilization of AFLP over the last decades. We emphasize that whenever the delimitation of evolutionary entities is the central goal, as it is in many fields of biodiversity research, AFLP is still an adequate technique.
Similar content being viewed by others


Introduction
Phylogeography has led to large advancements in understanding the spatio-temporal evolution of species and the underlying climatic, geological, and ecological processes1. Easy access to molecular-genetic data has propelled large-scale and cross-species phylogeographic studies and offered new insights on long-standing questions such as the postglacial colonization of Europe2. As such, phylogeography has become an integral part of biogeographic research in general3. Owing to the large effort necessary to obtain multiple, informative genetic markers from the genomes of non-model organisms, many studies had to rely on single or a few genetic markers in the past. As broadly discussed, studies utilizing a single or few genetic markers are affected by incongruences due to, for example, pseudogene amplification4, or simply different gene genealogies supporting alternative species trees5. As a consequence, many journals important to the field decided to no longer accept such studies (e.g. Molecular Ecology). Today, the phylogenetic resolution at different time scales and on different taxonomic levels that can be achieved via high-throughput sequencing (HTS) data is unprecedented6. The need to look for alternative ways to generate multi-locus datasets has been constantever since the dawn of molecular systematics.
An established and widely used method to generate genetic markers from any organism’s genome is amplified fragment length polymorphism (AFLP) fingerprinting7. This tool from the pre-HTS era is able to sample several hundred to a few thousand genetic markers from an organism without any prior knowledge of its genome. The method has been extensively used in the field of evolutionary ecology, spanning disciplines from phylogenetics to phylogeography and species delimitation8,9. AFLP has been employed for over 20 years, and its applicability has been proven in thousands of studies (Fig. 1A), including also recent publications in highly visible journals10,11,12,13.

Results of a literature survey on usage, funding, and taxonomic scope of amplified fragment length polymorphism (AFLP, blue bars) and restriction site associated DNA sequencing (RADseq, red bars) studies (details in Supplementary Methods, Supplementary Results). (A) Number of publications produced per year using AFLP and RADseq. Total number of studies published until the end of 2019: AFLP, 13,173; RADseq, 1230; introduction of the respective technique (AFLP: 1995; RADseq: 2008) (B) Number of publications using AFLP and RADseq grouped by nationality of the funding agency. (C) Number of phylogeographic publications using AFLP and RADseq grouped by taxonomic study-group.
The advancement of molecular methods in the era of HTS helped phylogeography, once data-limited, to become a data-rich field14. One HTS-based method, which has been particularly used for phylogeographic studies, is restriction site associated DNA sequencing (RADseq)15. This method is able to produce, depending on the experimental design, thousands to tens of thousands of gene fragments that can be used to infer large numbers of single nucleotide polymorphisms (SNPs) from populations of any non-model organism’s genome16. The generation of real sequence data that can be analyzed using established models of molecular evolution is a pivotal advantage of RADseq, compared with other marker types such as microsatellites and the above-mentioned AFLP fingerprints. Also, the method’s power in terms of resolution has been empirically demonstrated compared with genotyping methods such as microsatellites or multiple single-locus markers5,17,18,19,20.
Nevertheless, RADseq is akin to AFLP in terms of discovering markers throughout a genome by subsampling regions targeted by specific restriction enzymes15. Methodological limitations and features of both techniques have been thoroughly discussed, and parallels become evident, considering, for example, the problems both methods were reported to have in sampling markers from large, complex genomes21,22. Still, RADseq has clear advantages in terms of reproducibility and marker discovery rate23, and, as co-dominant sequence data instead of presence-absence data are generated, also a much wider range of applications16,24. The mere number of loci available, however, is not always important to answer a biological question. For example, it has been demonstrated that dozens of simple sequence repeats (SSRs) lead to the same result as hundreds to thousands of SNPs17,20,25. Such comparisons need to be interpreted carefully, especially if this is done in a phylogenetic context: while models for molecular evolution have been adapted and empirically tested for RADseq derived SNP data26, such models are under discussion for SSRs27, or not available in case of AFLP. While RADseq is definitely able to resolve fine-scale phylogeographic patterns via large numbers of SNPs5,28, the question of how many markers are really needed to do so, has not been answered so far.
A disadvantage of HTS techniques is their resource intensity. While the costs per sequenced base pair are constantly decreasing, a cost shift towards laboratory equipment and computational facilities has occurred that, in some setups, even counterbalances the cost-advantage of decreasing sequencing prices29,30. The overall costs depend on the HTS protocol used. Single-digest RADseq protocols, for example, rely on DNA shearing via focused ultra-sonication15, which calls for expensive sonicators that are not standard in most labs. Beside other options, one of the most frequently applied and reliable reduced-representation HTS techniques is double-digest RADseq (ddRADseq). In this case, DNA shearing is done enzymatically, which improves the tunability of fragment size selection. To ensure an accurate fragment-size selection, special electrophoresis devices are commonly used for ddRADseq31. However, such devices are generally much cheaper than sonicators, and ddRADseq has been emphasized to be more cost-efficient than single-digest RADseq31. Generally, large purchases can be avoided by outsourcing steps that depend on expensive machinery to the sequencing companies for additional service charges.
Resource imbalances around the globe, and therefore imbalances in the access to cutting-edge technologies like HTS do matter to biology. Some of the world’s economically most disadvantaged countries harbor the majority of the planet’s biodiversity but are disproportionately understudied, and local research is often chronically underfunded32,33,34. Funding for both RADseq and AFLP studies appears biased towards economically rich countries, as shown in Fig. 1B. This bias is less severe in the case of the AFLP technique, which is often used in newly industrialized economies, such as India or Brazil (Fig. 1B). Institutions and scientists based in economically disadvantaged regions will be pivotal in dealing with the biodiversity crisis35,36,37. In this context, phylogeography, a discipline that is essential in defining conservation-relevant evolutionary entities and in addressing the taxonomic impediment, will be key38,39,40.
The perceived neglect to study “what is really out there”41 is by no means restricted to economically disadvantaged regions. A significant taxonomic bias is observable in both AFLP- and RADseq-based phylogeographic studies (Fig. 1C), and very species-rich groups, such as fungi, annelids, or echinoderms, are severely under-represented (Fig. 1C). In this context, it is especially worthwhile to explore the possibility that a well-established, cheap, and reliable tool such as AFLP could ultimately produce results that are as robust as genomic SNPs.
Here, we present a direct comparison between AFLP and RADseq by applying these two methods to the same dataset comprising six species of plants and arthropods co-distributed in the Eurasian steppes42. In all six taxa, a separation into at least two distinct groups reflecting the complex historic biogeography of Eurasian steppes has been revealed using RADseq in a large-scale study of Kirschner, Zaveska et al. 202042. Specifically, we ask the following questions: (i) Do AFLP and RADseq retrieve similar phylogeographic patterns? Assuming that thousands of RADseq loci are more likely to resolve the phylogeographic structure of a species, we examine to what extent AFLP-based results reflect RADseq results. To evaluate possible differences between both methods, we applied phylogenetic network analysis and a set of similarity statistics. (ii) Does the information content in terms of phylogeographic information per locus of AFLP and RADseq results differ significantly? We evaluated the information content by resampling loci in a rarefaction approach for RADseq and by comparing them with AFLP markers. This allowed us to directly observe and compare the information content of the respective dataset. We will address both questions by applying our approach to six unrelated taxa with different evolutionary histories, genome sizes, ploidy levels, and spatiotemporal population dynamics. In addition, we produced a larger dataset for one of the six taxa for an in-depth analysis of the robustness of AFLP.
Results
Locus yield of AFLP and RADseq
After quality control, AFLP yielded between 100 and 600 fragments in the cross-taxonomic dataset containing all six taxa, and 985 for the large Omocestus petraeus dataset, containing only a single taxon (Table 1). The total number of RADseq loci in both datasets ranged from 5000 to 15,000 (Table 1).
RADseq and AFLP contained similar information in four out of six studied taxa
Information content of AFLP and RADseq markers was significantly correlated for Astragalus onobrychis, Euphorbia seguieriana, Plagiolepis taurica and O. petraeus (for both the cross-taxonomic dataset and the large O. petraeus dataset), as shown by the Mantel tests of intraspecific distance matrices derived from the respective datasets (Fig. 2C). In Stipa capillata and Stenobothrus nigromaculatus, however, this test showed no significant correlation (Fig. 2C). Therefore, comparative analyses concerning these two taxa were not interpreted any further.

Comparative analyses for the six study species based on amplified fragment length polymorphism (AFLP) and restriction site-associated DNA (RADseq) sequencing data Abbreviations in A-B reflect the geographic origin of the sample: A1 = Western Alps, A2 = Eastern Alps, B = southeastern Balkan, CA = Central Asia, P = Pannonia. (A) NeighborNets inferred from AFLP data and RADseq data. (B) Ordination of AFLP and RADseq data via non-metric multidimensional scaling (NMDS). (C) Correlation plots of AFLP and RADseq derived distance matrices. Mantel R and corresponding significance levels are shown in the plot. (D) Global R values derived from analysis of similarity (ANOSIM)79 of down-sampled RADseq matrices. Each point represents one randomly down-sampled observation; colored points are significant observations, grey points are non-significant. Larger points show calculations based on the number of available AFLP loci. (E) Correlation coefficient R calculated via Mantel matrix correlation of down-sampled RADseq matrices and the full RADseq matrix. Each point represents one randomly down-sampled observation; coloured points are significant observations, grey points are non-significant. Large red points show calculations based on the number of available AFLP loci.
The presence of similar information content of AFLP and RADseq datasets was further illustrated by NeighborNet topologies and NMDS (non-metric multidimensional scaling) ordinations (Fig. 2A-B, Fig. 3A,C), which resulted for the cross-taxonomic dataset in similar backbone patterns with minor deviations (e.g. clustering of Central Asian samples in O. petraeus, Fig. 2A); or in the case of the large O. petraeus dataset, in the same clustering pattern (Fig. 3A). Analysis of similarity (ANOSIM) based on the geographic location resulted in similar between-region dissimilarities when using AFLP and RADseq data; an exception was P. taurica, where the AFLP-derived Global R value was much lower (Table 1).

Comparative analyses based on the large Omocestus petraeus amplified fragment length polymorphism (AFLP) and restriction site associated DNA sequencing (RADseq) datasets. Abbreviations in A-C reflect the geographic origin of the samples: A1 = Western Alps, A2 = Eastern Alps, B = southeastern Balkan Peninsula, CA = Central Asia, P = Pannonia (A) NeighborNets inferred from AFLP data and RADseq data. (B) Bar plots show the two genetic groups identified via Bayesian clustering. Each bar represents one individual, colors show proportion of affiliation to a cluster. (C) Ordination plots of AFLP and RADseq data retrieved from non-metric multidimensional scaling (NMDS). (D) Correlation plots of AFLP and RADseq derived distance matrices. Mantel R and corresponding significance levels are shown in the plot. (E) Global R values calculated via analysis of similarity (ANOSIM) derived from the down-sampled RADseq matrices. Each point represents one randomly down-sampled observation; colored points are significant observations, grey points are non-significant. Large orange points show calculations based on the number of available AFLP loci. (F) Correlation coefficient R calculated via Mantel matrix correlation of the down-sampled RADseq matrices and the full RADseq matrix. Each point represents one randomly down-sampled observation; colored points are significant observations, grey points are non-significant. Larger points show calculations based on the number of available AFLP loci.
The largest similarity in pattern and information content was observed between the large O. petraeus AFLP and RADseq datasets (Table 1, Fig. 3A,C,D). For these data, also Bayesian clustering analysis resolved two identical clusters irrespective of the data used (Fig. 3B, Supplementary Fig. 1).
AFLP matched RADseq derived parameters under rarefaction in two out of six taxa
Random down-sampling of RADseq loci showed that ANOSIM-derived Global R values approached values inferred from the complete RADseq dataset when at least 2000 RADseq loci were subsampled from the original dataset (Fig. 2D). In some instances, even a lower number of loci led to similar results; however, smaller subsamples, led to large deviations (Fig. 2D). The largest deviations were observed in downsampling the small O. petraeus dataset. Here, Global R values were not fully converging, even with the full loci number as data basis (Fig. 2D). This effect was also observed in the down-sampled large O. petraeus dataset (Fig. 3E, Table 1). In the latter case, ANOSIM analysis was not even applicable when less than 1250 loci were subsampled due to the large quantity of missing data in the SNP matrix (Fig. 3E). Global R values calculated from AFLP data from A. onobrychis and O. petraeus (cross-taxonomic and large datasets) reached and even exceeded the Global R values of the corresponding downsampled RADseq dataset. However, in the case of P. taurica and E. seguieriana, the Global R values derived from AFLP did not reach the respective values obtained with the RADseq dataset.
For both plant species, Mantel correlation coefficients R estimated from the downsampled SNP matrices showed that full matrix similarity (R = 1) could be reached with approximately 1000 randomly-drawn RADseq loci (Fig. 2E). In the case of P. taurica, about 2000 loci were necessary to achieve this correlation coefficient, while for O. petraeus, matrix similarity was only gradually reached (cross-taxonomic & large datasets) (Fig. 2E). When AFLP distance matrices were tested for correlation with the corresponding downsampled RADseq based distance matrices, comparable and even higher matrix similarity coefficients were obtained in A. onobrychis and O. petraeus (Fig. 2D, Fig. 3F, Table 1). In E. seguieriana and P. taurica, the downsampled RADseq and AFLP Mantel correlation coefficients were smaller.
The amount of missing data in the full RADseq SNP matrices was randomly distributed after initial filtering and did not change in the performed random downsampling analyses.
Discussion
We show that AFLP and RADseq-derived genomic data can yield similar phylogeographic patterns. Specifically, we employed a comparative phylogeographic framework to compare AFLP and RADseq datasets from six plant and animal species in terms of statistical similarity of phylogeographic patterns and information content. In four out of the six taxa, these two dataset-types were statistically similar in information content and resulted in nearly identical phylogeographic patterns (Figs. 2, 3). Surprisingly, a smaller dataset of AFLP loci (157 to 985) was akin to thousands of RADseq loci (5946 to 13,335) in their ability to resolve intraspecific genealogical patterns in these taxa. Compared with the RADseq-inferred results as benchmark, the robustness of AFLP based results in terms of information content and phylogeographic resolution is even more remarkable, especially when taking into account the distinct genome sizes (Supplementary Table 1), ploidy levels (diploid, tetraploid and octoploid populations in A. onobrychis43), evolutionary histories (taxa from two kingdoms and five different families) and spatiotemporal dynamics of the studied species.
In two species, however, AFLP loci failed to provide interpretable results. It might be that the locus yield (603 loci for the grasshopper S. nigromaculatus and 157 for the grass S. capillata) provided insufficient resolution, a point supported by the shallow phylogeographic structure detected with RADseq. We suspect that in the case of S. nigromaculatus the large size of the genome (11.36 giga base pairs) was responsible for the failure. An alternative explanation applicable to both species is the disproportionally high occurrence of repetitive elements in the genome, which has been reported for other grasses and Caelifera grasshoppers44,45. On the other hand, AFLPs have been able to resolve phylogeographic patterns in other studies of S. capillata, so a mere methodological issue in this study cannot be ruled out as error source46. However, this example highlights the limitations associated with the use of non-sequence-based genetic loci, such as the difficulties to infer the reasons behind method failure. It also highlights the importance of gathering information on biologically-dependent factors, such as genome size, especially when working with non-model organisms. If large genomes or a high proportion of repetitive elements in the genome are expected, scientists should refrain from using AFLP in favor of other techniques, such as RADseq. Because of these reasons, results from S. capillata and S. nigromaculatus are excluded from the discussion below.
Our results showed that AFLP performed equally well as RADseq when comparing the information content of dissimilarity matrices with regard to phylogeographic patterns (Global R) in two species (A. onobrychis, O. petraeus; Fig. 3D). Similarity of intraspecific dissimilarity matrices, however, did not increase when comparing downsampled RADseq datasets with AFLP (Fig. 2E). The downsampling revealed that in most cases fewer than 1000 RADseq loci were sufficient to reach the Global R of the full dataset. This indicates that in many cases a fraction of the SNP dataset might be sufficient to infer the phylogeographic structure within a species.
Generating an additional dataset for one species (O. petraeus), containing five times more individuals than the small cross-taxonomic dataset, enabled us to evaluate the influence of sample size on AFLP locus yield and, hence, the dataset information content. Compared with the cross-taxonomic dataset, the large O. petraeus dataset contained more AFLP loci (cross-taxonomic dataset: 486; large dataset: 985), and both dataset similarity and information content increased when compared with the corresponding large RADseq dataset (Fig. 3E–F, Table 1). As a consequence, the phylogenetic resolution of the large AFLP dataset was also better than in the cross-taxonomic AFLP dataset, and the incongruence observed in the latter (i.e. the clustering of some Central Asian individuals, Fig. 2A) disappeared (Fig. 3A). The downsampled AFLP dataset resulted in an even stronger support for the defined groups than the RADseq dataset (Global R, Fig. 3E).
A correlation between sample size (i.e. number of sampled populations per region) and phylogenetic resolution has been demonstrated in other RADseq-based phylogenetic studies47,48 and for several AFLP-inferred population genetic measures49. Small sample sizes and missing data might not be as much a problem for RADseq as for AFLP. Despite the large amount of missing data in the O. petraeus RADseq datasets, which hindered the calculation of Global R values from the downsampled data (Fig. 3E), the phylogeographic structure recovered by both RADseq datasets was similar, but this was not true for AFLP data (Figs. 2A, 3A). Given this, we emphasize here the need to include as many populations from the studied units (e.g. populations, regions) as feasible when using the AFLP technique. Comparing only a few populations is not only prone to produce ambiguous results but also limits data analyses. For instance, while we encountered convergence problems when analyzing the cross-taxonomic AFLP datasets with STRUCTURE50 (not shown), the same method worked well when applied to the large O. petraeus dataset and resulted in a similar clustering pattern, irrespective of the data type used (K = 2, Fig. 3B, Supplementary Fig. 1). However, the admixture observed in the AFLP-based clustering, and the lack of such signal in the RADseq based clustering, likely reflected noise in AFLP data rather than a true admixture signal. Similar to previous studies49, we found that small sample sizes can lead to ambiguous or erroneous results when inferring population structure from AFLP data.
While AFLP proved to be able to resolve intraspecific phylogenetic relationships in the study species, it is important to bear in mind that the phylogenetic methods utilized here are solely distance-based. In more complex scenarios, where large evolutionary distances between species and, consequently, large amounts of homoplasy are expected, distance-based phylogenetic methods in general, and the usage of AFLP in particular, are problematic51. In such scenarios, phylogenetic methods with underlying mechanistic models of molecular evolution, as implemented for example in most likelihood based approaches52, would be needed to adequately resolve phylogenetic relationships53. Such models of molecular evolution have been developed and extensively tested over decades of phylogenetic research and are easily applicable to DNA sequence data53 and, with some restrictions, to SNP data26,54, but not to AFLP. However, we want to point out that distance-based methods are solid options at the intraspecific or population level with low rates of overall change, which is the case in this study.
We emphasize that the intention of our study is not to advocate the use of allegedly old-fashioned alternatives to HTS techniques. On the contrary, we are convinced that the latter have revolutionized and will further revolutionize biology. It might be just a matter of time until these techniques will finally replace most “traditional” techniques that are still around, even if some of them, such as microsatellites, have shown to be remarkably resilient in competition against high-throughput sequencing techniques55. It is, however, unrealistic that the global scientific community is able to keep up in terms of methodology with economically rich labs working, for example, on questions in population and speciation genomics that require large sequencing depth and high-end bioinformatic analyses. While these players are perceived to constantly push the frontier, we should keep in mind that a substantial part of the theoretical basis of today’s population genomics originates from pre-sequencing days. Hence, the methodological toolbox available to a scientist can and should not be seen as a decisive factor to get papers published or projects funded, as long as the method applies to common standards, such as reproducibility. We think this is particularly important to bear in mind if we aim to extend the narrow taxonomic focus of current phylogeographic studies (Fig. 1C).
The methodological advancements around sequencing techniques are very dynamic, and methods praised until recently, such as RADseq, have also been criticized for their limitations, and might eventually be replaced by other techniques soon56,57. Given the pace of methodological developments that is obviously able to rapidly render methods obsolete, we highlight two points that we consider important. First, we showed that in a comparative phylogeographic scenario, meaningful phylogeographic patterns that were inferred via AFLP, an over 20-year old technique, survived a direct comparison with results inferred from RADseq. While the emphasized analytical and methodological limitations need to be considered, we show that AFLP data are robust and reliable. This is important concerning the backward compatibility of AFLP data in terms of significance of discoveries that have been made via this technique in the past. We emphasize that scientists that are successfully using this marker system in their lab, still have AFLP datasets, or simply cannot afford to switch to HTS methods right now should not be discouraged from using AFLP if the method is adequate to address the biological question asked. Secondly, many urging biological questions can be solved with simple methods. In other words, it might be unnecessary to obtain thousands of SNPs if a method like AFLP is sufficient to, for example, delimit phenotypically cryptic entities58, detect hybrid speciation11, or infer large scale phylogeography12. Facing the ongoing biodiversity and climate crisis, conservation policy-makers will need quick, large-scale, and straightforward answers59. To address this challenge, it will be crucial to strengthen the link of conservation genetics and conservation practice60. Thus, we want to emphasize that any source of robust molecular evidence should be seized in doing so.
In many scenarios, financial and personnel resource limitations are important arguments in favor of adapting the employed methodology to the specific question and not vice versa. As shown in this study, it is possible to robustly infer phylogeographic patterns by using AFLP. While advantages of high-throughput sequencing based methods like RADseq are obvious, we still want to encourage scientists and also publishers to maintain a critical stance towards the rampant method-centrism in an era of rapid methodological progress. Ultimately, the relevance of a result should be valued for its biological significance rather than the fanciness of the technique that was used to obtain it.
Material and methods
Taxon sampling and sample selection
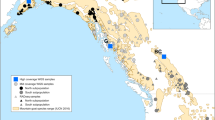
To cover a variety of evolutionary histories, three nominal species from three families of angiosperms, Astragalus onobrychis L. (Fabaceae), Euphorbia seguieriana Neck. (Euphorbiaceae), and Stipa capillata L. (Poaceae), and three arthropod species from two families, Plagiolepis taurica Santschi, 1920; (Formicidae), Omocestus petraeus (Brisout de Barneville, 1856), and Stenobothrus nigromaculatus (Herrich-Schäffer, 1840; both Acrididae), were selected. Samples of all taxa were collected between 2014 and 2016 from dry grassland localities from five regions, the Western Alps (A1), the Eastern Alps (A2), the Pannonian Basin (P), the southeastern Balkan Peninsula (B), and Central Asia (CA) (Fig. 4), in the course of a project to study the phylogeography of the Eurasian steppe biome42. Specimens were hand sampled and stored in silica gel (plants) or 96% ethanol (animals) for further analyses. For comparative analyses, distances among sampling localities for each taxon within the same region were always below 20 km, except the sampling localities of S. nigromaculatus in the Balkan Peninsula, which spanned 150 km. Three individuals per taxon and region were selected, resulting in a “cross-taxonomic dataset” of 90 samples for AFLP and RADseq analyses. In addition to this cross-taxonomic dataset, a second, single-taxon ”large dataset” was generated for O. petraeus. The latter comprised 81 individuals, evenly sampled from the five regions mentioned above.

Map of Europe and Central Asia showing the sampling regions; A1 = Western Alps, A2 = Eastern Alps, B = southeastern Balkan Peninsula, CA = Central Asia, P = Pannonia. The underlying map is based on the GTOPO30 Global Digital Elevation Model (available at the United States Geological Service EROS Data Center, https://doi.org/10.5066/F7DF6PQS) and has been modified using QGIS version 3.10.5 (https://qgis.org/).
DNA extraction
Plant DNA was extracted from leaf tissue using a sorbitol/high-salt cetyltrimethylammonium bromide method61. The extract was purified using the NucleoSpin gDNA clean-up kit (Macherey–Nagel, Düren, Germany). Animal DNA was extracted from leg tissue (O. petraeus and S. nigromaculatus) or whole animals (P. taurica) with the DNeasy Blood & Tissue Kit (Qiagen, Düsseldorf, Germany). The same individual extracts were used for AFLP and RADseq experiments except for Plagiolepis, where extracts of two separate individuals from each nest had to be used due to the low DNA yield from a single individual.
Amplified fragment length polymorphism fingerprinting
The AFLP protocol used for this study is described in detail in Wachter et. al.62. Briefly, DNA samples of all individuals were digested using the restriction enzymes MseI and EcoRI. Three randomly selected samples per species were added as "blind" samples to test for reproducibility and contamination, resulting in 18 replicated samples. For the large dataset, three replicates were randomly selected from each region, resulting in 15 replicated samples. Restriction digestion and ligation of the adapters was followed by pre-selective PCR amplification. The cycling conditions were: 2 min at 72 °C, followed by 30 cycles of 30 s at 94 °C, 30 s at 56 °C, and 2 min at 72 °C, and a final extension step of 10 min at 60°C62. For selective amplification, eight primer combinations (tEco-ACA/Mse-CAC; tEco-ACT/Mse-CTC; tEco-ACA/Mse-CAT; tEco-ATC/Mse-CTG; tEco-ACC/Mse-CAG; tEco-ACC/Mse-CAT; tEco-AAC/Mse-CAT; and tEco-AGC/Mse-CTG) were used, with each forward primer having a 5′ M13 tail (t). The cycling scheme was: 2 min at 94 °C, followed by 13 cycles of 30 s at 94 °C, 30 s at 65 °C decreased by 0.7 °C / cycle and 2 min at 72 °C, followed by 24 cycles 30 s at 94 °C, 30 s at 56 °C, and 2 min at 72 °C, completed by a final extension step of 10 min at 72°C58. FAM / HEX / NED / PET labelled M13 primers were added in the ratio M13:F:R = 10:1:10. Fragment analysis was performed by the Comprehensive Cancer Center DNA Sequencing & Genotyping Facility (University of Chicago, USA) on an ABI 3730 sequencer (Applied Biosystems, Chicago, USA).
Scoring and quality assessment of AFLP markers
AFLP profiles were converted using Peakscanner v.1.0 (Applied Biosystems). Subsequently, optiFLP v.1.5163 was used in unsupervised mode to identify optimal parameters for scoring. The final peak scoring using the inferred parameters was done in tinyFLP v.1.4064. The three randomly selected replicates were used to assess whether a single AFLP locus carried correct biological information: in theory, the distance between these biological replicates should be zero (i.e., they should have identical AFLP profiles). In practice, a zero distance was rarely achieved, due to various factors introducing noise in peak scoring. When removal of a single locus from the binary AFLP matrix reduced the genetic distance between the replicated samples, this locus was considered to be affected by noise. In accordance with these considerations, a custom Python script (Supplementary Material) was used for the following procedure. First, the sum of p-distances between all pairs of replicated samples was determined. Then, the first locus was removed from the AFLP matrix, and the sum of distances was calculated again and compared with that of the full matrix. When the p-distance after locus removal was lower than in the full matrix, this locus was removed; otherwise the matrix remained unchanged. The same test was repeated for each locus. The script generated a new matrix file and a logfile with information on which loci were removed and how the sum of p-distances changed.
The R package vegan65 was used to calculate intraspecific distance matrices using Jaccard distances and visualize these matrices via non-metric multidimensional scaling (NMDS) adding 90% confidence interval ellipses. Based on these ordinations, individuals and/or primer combinations were excluded if an individual appeared outside the confidence interval in more than 50% of all primer combinations and if more than 10% of all individuals were outside the interval in a single primer combination.
Restriction site associated DNA sequencing
Each taxon’s relative genome size was determined by flow cytometry66, whereby leaf tissue (plants), leg muscle tissue (O. petraeus and S. nigromaculatus), and whole heads (P. taurica) were used. Given the genome sizes, the desired sequencing depth and total fragment yield, the number of individually barcoded samples that could be pooled into a single RADseq library, and the optimal restriction enzyme for each taxon were assessed via RADseq counter67. RADseq libraries were prepared using a protocol modified from Paun et al.68. Per individual, 250 ng DNA (40 ng for P. taurica) was used for restriction digestion with the enzymes SbfI (O. petraeus and S. nigromaculatus) and PstI (plants and P. taurica). A double barcoding approach was chosen to decrease the number of adapters necessary to pool 96 individuals into a single library. A six-base-pair (bp) P2 barcode and an eight-bp P1 barcode, each differing by at least two bases from the respective barcodes belonging to the same adapter category, were selected to avoid erroneous assignment of fragments. P1 adapters (200 mM) were ligated to the restricted samples overnight at 16 °C. Samples were sheared in a two-minute-long, focused ultrasonication program using a sonicator (M220 series, Covaris Inc., Woburn, USA) to obtain average fragment lengths of 400 bp. To remove undesired fragment lengths from each pool, left- and right-side size selection steps were carried out, using × 0.7 and × 0.55 volume of SPRIselect reagent (Beckman Coulter, California, USA). After ligation of P2 adapters, samples were pooled. Before, the DNA content of each sample was quantified with a fluorometer (Fluoroskan Ascent, Thermo Scientific, Schwerte, Germany) using a fluorescent dye (Invitrogen Quant-iT PicoGreen dsDNA Assay Kit, Thermo Scientific, Schwerte, Germany). Samples were diluted to be equally represented in the final pool. Additional size selection steps were conducted on the left side of the target range using × 0.55 volume of SPRI reagent before and after the 18 cycles of PCR amplification with Phusion Master Mix (Thermo Fisher Scientific, Schwerte, Germany). The libraries were sequenced on a HiSeq2000 sequencer (Illumina, San Diego, United States) at the Vienna BioCenter (https://www.viennabiocenter.org/facilities/next-generation-sequencing/) as 100-bp single end reads.
Identification of RADseq loci and SNP calling
Illumina raw reads were quality filtered and demultiplexed via the program process_radtags.pl, and RADseq tag catalogs were assembled and SNPs were called using the denovo_map.pl pipeline implemented in Stacks v. 1.4669. Large genomes, as observed in O. petraeus and S. nigromaculatus, are prone to contain large proportions of pseudo-genes, transposable elements and non-coding DNA45. To exclude such regions from the analyses, RepeatMasker70 was used to identify and mask repeated elements in the Locusta migratoria genome45 (GenBank: AVCP000000000.1), and the quality-filtered reads of Omocestus and Stenobothrus were then mapped to the masked L. migratoria genome using Stampy v1.0.2071. Only the raw reads that mapped on the masked L. migratoria genome were included in the final dataset.
Populations.pl69 was used to export SNP matrices in STRUCTURE50 format and phylip format72. Whitelists were used to exclude fragments with more than 10 SNPs, deleveraged stacks, and loci with more than 75% missing calls (85% in case of O. petraeus). To avoid violation of the assumptions of site-independent models developed for analysis of unlinked SNP data, we selected one random SNP per RADseq fragment using the write_random_snp flag for the Bayesian clustering analysis in STRUCTURE50. Phylip files were generated with the phylip_var option, which adds variant sites to the phylip output using IUPAC notation.
Bayesian clustering analysis
Bayesian cluster analyses were done in the software STRUCTURE v.2.3.4.50. STRUCTURE runs for K = 1 to K = 5 were conducted, reflecting the number of sampled regions, with 10 replicates for each K using the default settings. Each MCMC ran for 2,000,000 generations, and the first 200,000 generations were discarded as burn-in. Bar plots and likelihood graphs were generated by the software CLUMPP v1.1.273 and distruct v1.174. The optimal number of clusters was determined following Evanno et al.75.
Phylogenetic analyses and analyses based on intraspecific dissimilarity matrices
Phylip files based on RADseq data generated via populations.pl69, and 0/1 matrices based on AFLP data were used to calculate NeighborNets in SplitsTree4 v4.14.476 using standard settings. NeighborNets were preferred over bifurcating neighbor joining trees, as networks provide a more complete picture concerning the pattern and the uncertainty of the retrieved splits.
The same matrices were used to calculate intraspecific dissimilarity matrices in the R package vegan65. To infer these distances within each dataset, Jaccard dissimilarities were used for AFLP data, and Gower distances for RADseq SNP data, as suggested for each data type77,78. For each species we were trying to assess (i) the similarity of AFLP and RADseq data and (ii) the role of geographic distance in the structuring of genetic variance among and within populations. To assess the first point (i) intraspecific distance matrices obtained from the AFLP and RADseq data were tested for correlation with Mantel tests, using the Pearson correlation method and 9999 random permutations in vegan65. To explore the second point (ii), the same R package was used to conduct Analysis of Similarity (ANOSIM)79, using geography (i.e. the sample region) as prior. Global R values returned by this analysis describe similarity within the defined populations and dissimilarity among them. Values close to 1 suggest similarity within populations and dissimilarity among them, while a value close to 0 indicates a lack of geographic structure among populations79. Finally, distance matrices were plotted in non-metric space via non-metric multidimensional scaling (NMDS). This was done in the R package vegan using the function that exhaustively iterates the scaling process until an optimal solution is reached65.
Rarefaction
A random locus resampling approach was developed to assess the information content (i.e. how many loci are sufficient to obtain results similar to the full dataset) of RADseq data with decreasing locus number. Random resampling was done on modified SNP matrices exported via the structure flag in populations.pl69. Using custom R scripts, loci were sampled from the original SNP matrix in steps of 50 loci and 50 replicates per step, without sampling any locus twice in the same replicate. At each step, global R was inferred via ANOSIM, and a Mantel correlation test of the sub-sampled matrix with the full dataset was performed. The Global R and correlation coefficient R (Mantel test) obtained at each step were plotted using ggplot280. Lines connecting the mean of each rarefaction step and the respective standard deviations were added to the plot. Global R and correlation coefficient R calculated from the corresponding AFLP dataset were added to the plots to compare the results with a correspondingly down-sampled RADseq dataset.
Data Availability
RADseq data are available via the NCBI Sequence Read Archive (BioProject ID PRJNA680892). AFLP data are available in tabular format in Supplementary Data 1.
References
Avise, J. C. Phylogeography: retrospect and prospect. J. Biogeogr. 36, 3–15 (2009).
Hewitt, G. M. Post-glacial re-colonization of European biota. Biol. J. Linn. Soc. 68, 87–112 (1999).
Linder, P. H. Phylogeography. J. Biogeogr. 44, 243–244 (2017).
Song, H., Buhay, J. E., Whiting, M. F. & Crandall, K. A. Many species in one: DNA barcoding overestimates the number of species when nuclear mitochondrial pseudogenes are coamplified. Proc. Natl. Acad. Sci. 105, 13486–13491 (2008).
Philippe, H. et al. Pitfalls in supermatrix phylogenomics. Pitfalls supermatrix phylogenomics. Eur. J. Taxon. 28, 3. https://doi.org/10.5852/ejt.2017.283 (2017).
Villaverde, T. et al. Bridging the micro- and macroevolutionary levels in phylogenomics: Hyb-Seq solves relationships from populations to species and above. New Phytol. 220, 636–650 (2018).
Vos, P. et al. AFLP: A new technique for DNA fingerprinting. Nucleic Acids Res. 23, 4407–4414 (1995).
Meudt, H. M. & Clarke, A. C. Almost forgotten or latest practice? AFLP applications, analyses and advances. Trends Plant Sci. 12, 106–117 (2007).
Paun, O. & Schönswetter, P. Amplified fragment length polymorphism: an invaluable fingerprinting technique for genomic, transcriptomic, and epigenetic studies. Methods Mol. Biol. 862, 75–87 (2012).
Dejaco, T., Gassner, M., Arthofer, W., Schlick-Steiner, B. C. & Steiner, F. M. Taxonomist’s nightmare … evolutionist’s delight: an integrative approach resolves species limits in jumping bristletails despite widespread hybridization and parthenogenesis. Syst. Biol. 65, 947–974 (2016).
Sefc, K. M. et al. Shifting barriers and phenotypic diversification by hybridisation. Ecol. Lett. 20, 651–662 (2017).
Suchan, T., Malicki, M. & Ronikier, M. Relict populations and Central European glacial refugia: the case of Rhododendron ferrugineum (Ericaceae). J. Biogeogr. 46, 392–404 (2019).
Schneeweiss, G. M. & Schönswetter, P. A re-appraisal of nunatak survival in arctic-alpine phylogeography. Mol. Ecol. 20, 190–192 (2011).
Lemmon, A. R. & Lemmon, E. M. High-throughput identification of informative nuclear loci for shallow-scale phylogenetics and phylogeography. Syst. Biol. 61, 745–761 (2012).
Baird, N. A. et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS ONE 3, 1–7 (2008).
Andrews, K. R., Good, J. M., Miller, M. R., Luikart, G. & Hohenlohe, P. A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 17, 81–92 (2016).
Jeffries, D. L. et al. Comparing RADseq and microsatellites to infer complex phylogeographic patterns, an empirical perspective in the Crucian carp, Carassius carassius L.. Mol. Ecol. 25, 2997–3018 (2016).
Bohling, J., Small, M., Von Bargen, J., Louden, A. & DeHaan, P. Comparing inferences derived from microsatellite and RADseq datasets: a case study involving threatened bull trout. Conserv. Genet. 20, 329–342 (2019).
Lemopoulos, A. et al. Comparing RADseq and microsatellites for estimating genetic diversity and relatedness—implications for brown trout conservation. Ecol. Evol. 9, 2106–2120 (2019).
Mesak, F., Tatarenkov, A., Earley, R. L. & Avise, J. C. Hundreds of SNPs vs. dozens of SSRs: which dataset better characterizes natural clonal lineages in a self-fertilizing fish?. Front. Ecol. Evol. 2, 74 (2014).
Fay, M. F., Cowan, R. S. & Leitch, I. J. The effects of nuclear DNA content (C-value) on the quality and utility of AFLP fingerprints. Ann. Bot. 95, 237–246 (2005).
Karam, M.-J., Lefèvre, F., Dagher-Kharrat, M. B., Pinosio, S. & Vendramin, G. G. Genomic exploration and molecular marker development in a large and complex conifer genome using RADseq and mRNAseq. Mol. Ecol. Resour. 15, 601–612 (2015).
Etter, P. D., Bassham, S., Hohenlohe, P. A., Johnson, E. A. & Cresko, W. A. SNP Discovery and Genotyping for Evolutionary Genetics Using RAD Sequencing. Methods in Molecular Biology (Clifton, N.J.) Vol. 772, 157–178 (Springer, Berlin, 2011).
Davey, J. L. & Blaxter, M. W. RADseq: next-generation population genetics. Brief. Funct. Genomics 9, 416–423 (2010).
Głowacka, K. et al. Genetic variation in Miscanthus × giganteus and the importance of estimating genetic distance thresholds for differentiating clones. GCB Bioenergy 7, 386–404 (2015).
Leaché, A. D., Banbury, B. L., Felsenstein, J., De Oca, A. N. M. & Stamatakis, A. Short tree, long tree, right tree, wrong tree: new acquisition bias corrections for inferring SNP phylogenies. Syst. Biol. 64, 1032–1047 (2015).
Wu, C.-H. & Drummond, A. J. Joint inference of microsatellite mutation models, population history and genealogies using transdimensional Markov Chain Monte Carlo. Genetics 188, 151–164 (2011).
Emerson, K. J. et al. Resolving postglacial phylogeography using high-throughput sequencing. Proc. Natl. Acad. Sci. 107, 16196–16200 (2010).
Sboner, A., Mu, X., Greenbaum, D., Auerbach, R. K. & Gerstein, M. B. The real cost of sequencing: higher than you think!. Genome Biol. 12, 125 (2011).
Muir, P. et al. The real cost of sequencing: scaling computation to keep pace with data generation. Genome Biol. 17, 53 (2016).
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S. & Hoekstra, H. E. Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS ONE 7, e37135 (2012).
Mittermeier, R. A. & Mittermeier, C. G. Megadiversity: Earth’s Biologically Wealthiest Nations. in 501 (CEMEX, 1997).
Trimble, M. J. & van Aarde, R. J. Geographical and taxonomic biases in research on biodiversity in human-modified landscapes. Ecosphere 3, art119 (2012).
Waldron, A. et al. Targeting global conservation funding to limit immediate biodiversity declines. Proc. Natl. Acad. Sci. USA 110, 12144–12148 (2013).
Adenle, A. et al. Stakeholder visions for biodiversity conservation in developing countries. Sustainability 7, 271–293 (2014).
Adenle, A. A., Stevens, C. & Bridgewater, P. Global conservation and management of biodiversity in developing countries: an opportunity for a new approach. Environ. Sci. Policy 45, 104–108 (2015).
Barber, P. H. et al. Advancing biodiversity research in developing countries: the need for changing paradigms. Bull. Mar. Sci. 90, 187–210 (2014).
Byrne, M. Phylogeography provides an evolutionary context for the conservation of a diverse and ancient flora. Aust. J. Bot. 55, 316 (2007).
Dufresnes, C. et al. Conservation phylogeography: does historical diversity contribute to regional vulnerability in European tree frogs (Hyla arborea)?. Mol. Ecol. 22, 5669–5684 (2013).
Coates, D. J., Byrne, M. & Moritz, C. Genetic diversity and conservation units: dealing with the species-population continuum in the age of genomics. Front. Ecol. Evol. 6, 165 (2018).
Trimble, M. J. & van Aarde, R. J. Species inequality in scientific study. Conserv. Biol. 24, 886–890 (2010).
Kirschner, P. et al. Long-term isolation of European steppe outposts boosts the biome’s conservation value. Nat. Commun. 11, 1–10 (2020).
Záveská, E. et al. Multiple auto- and allopolyploidisations marked the Pleistocene history of the widespread Eurasian steppe plant Astragalus onobrychis (Fabaceae). Mol. Phylogenet. Evol. https://doi.org/10.1016/J.YMPEV.2019.106572 (2019).
Luo, M.-C. et al. Genome sequence of the progenitor of the wheat D genome Aegilops tauschii. Nature 551, 498–502 (2017).
Wang, X. X. et al. The locust genome provides insight into swarm formation and long-distance flight. Nat. Commun. 5, 2957 (2014).
Hensen, I. et al. Low genetic variability and strong differentiation among isolated populations of the rare steppe grass Stipa capillata L. Central Europe. Plant Biol. 12, 526–536 (2010).
Huang, H. & Knowles, L. L. Unforeseen consequences of excluding missing data from next-generation sequences: simulation study of RAD sequences. Syst. Biol 65, 1–9 (2014).
Crotti, M., Barratt, C. D., Loader, S. P., Gower, D. J. & Streicher, J. W. Causes and analytical impacts of missing data in RADseq phylogenetics: insights from an African frog (Afrixalus). Zool. Scr. 48, 157–167 (2019).
Sinclair, E. A. & Hobbs, R. J. Sample size effects on estimates of population genetic structure: implications for ecological restoration. Restor. Ecol. 17, 837–844 (2009).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Althoff, D. M., Gitzendanner, M. A. & Segraves, K. A. The utility of amplified fragment length polymorphisms in phylogenetics: a comparison of homology within and between genomes. Syst. Biol. 56, 477–484 (2007).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Felsenstein, J. Inferring Phylogenies (Oxford University Press Inc., Oxford, 2004).
Eaton, D. A. R., Spriggs, E. L., Park, B. & Donoghue, M. J. Misconceptions on missing data in RAD-seq phylogenetics with a deep-scale example from flowering plants. Syst. Biol. 66, 399–412 (2016).
Hodel, R. G. J. et al. The report of my death was an exaggeration: a review for researchers using microsatellites in the 21st century. Appl. Plant Sci. 4, 1600025 (2016).
Puritz, J. B. et al. Demystifying the RAD fad. Mol. Ecol. 23, 5937–5942 (2014).
Lowry, D. B. et al. Breaking RAD: an evaluation of the utility of restriction site-associated DNA sequencing for genome scans of adaptation. Mol. Ecol. Resour. 17, 142–152 (2017).
Wagner, H. C. et al. Light at the end of the tunnel: Integrative taxonomy delimits cryptic species in the Tetramorium caespitum complex (Hymenoptera: Formicidae). Myrmecol. News 25, 95–129 (2017).
Wheeler, Q. D. Taxonomic Shock and Awe. In The New Taxonomy (ed. Wheeler, Q. D.) 211–226 (CRC Press, Boca Raton, FL, 2008). https://doi.org/10.1201/9781420008562.ch10.
Holderegger, R. et al. Conservation genetics: linking science with practice. Mol. Ecol. 28, 3848–3856 (2019).
Tel-Zur, N., Abbo, S., Myslabodski, D. & Mizrahi, Y. Modified CTAB procedure for DNA isolation from epiphytic cacti of the genera Hylocereus and Selenicereus (Cactaceae). Plant Mol. Biol. Rep. 17, 249–254 (1999).
Wachter, G. A. et al. Pleistocene survival on central Alpine nunataks: genetic evidence from the jumping bristletail Machilis pallida. Mol. Ecol. 21, 4983–4995 (2012).
Arthofer, W., Schlick-Steiner, B. C. & Steiner, F. M. optiFLP: software for automated optimization of amplified fragment length polymorphism scoring parameters. Mol. Ecol. Resour. 11, 1113–1118 (2011).
Arthofer, W. TinyFLP and tinyCAT: software for automatic peak selection and scoring of AFLP data tables. Mol. Ecol. Resour. 10, 385–388 (2010).
Oksanen, J., Guillaume Blanchet, F., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., Minchin, P.R., O'Hara, R.B., Simpson, G.L., Solymos, P., Stevens, M.H.H., Szoecs, E. & Wagner, H. Vegan: Community Ecology Package. R package. (2017).
Doležel, J., Greilhuber, J. & Suda, J. Estimation of nuclear DNA content in plants using flow cytometry. Nat. Protoc. 2, 2233–2244 (2007).
Davey, F. & RADseq counter. (2012). https://www.wiki.ed.ac.uk/display/RADSequencing/Home. (Accessed: 15th June 2014)
Paun, O. et al. Processes driving the adaptive radiation of a tropical tree (Diospyros, Ebenaceae) in New Caledonia, a biodiversity hotspot. Syst. Biol. 65, 212–227 (2016).
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A. & Cresko, W. A. Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140 (2013).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. http://www.repeatmasker.org. (Accessed: 1st September 2016)
Lunter, G. & Goodson, M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 21, 936–939 (2011).
Felsenstein, J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol. 17, 368–376 (1981).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806 (2007).
Rosenberg, N. A. DISTRUCT: a program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138 (2004).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Huson, D. H. & Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2006).
Kosman, E. & Leonard, K. J. Similarity coefficients for molecular markers in studies of genetic relationships between individuals for haploid, diploid, and polyploid species. Mol. Ecol. 14, 415–424 (2005).
Miclaus, K., Wolfinger, R. & Czika, W. SNP selection and multidimensional scaling to quantify population structure. Genet. Epidemiol. 33, 488–496 (2009).
Clarke, K. R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 18, 117–143 (1993).
Wickham, H. ggplot2 (Springer, Berlin, 2009). https://doi.org/10.1007/978-0-387-98141-3.
Acknowledgements
We thank all members of the Research Group Molecular Ecology and the Research Group Evolutionary Systematics, both University of Innsbruck, for valuable discussion; P. Andesner, M. Magauer, and D. Pirkebner for help with laboratory work; and Oliver Hawlitschek and two anonymous reviewers for their comments on an earlier version of the manuscript.
Funding
The present study was co-funded by the Austrian Science Fund (FWF, project P25955 “Origin of steppe flora and fauna in inner-Alpine dry valleys” to P.S.), and the Tiroler Wissenschaftsfonds (TWF, UNI-0404/2066, “Comparing information efficiency of high- versus low-resolution genome scans for phylogeographic studies” to P.K.). The computational results presented have been achieved using the HPC infrastructure LEO of the University of Innsbruck.
Author information
Authors and Affiliations
Author notes
A comprehensive list of consortium members appears at the end of the paper.
Consortia
Contributions
W.A., B.C.S.-S. and F.M.S. planned and designed the study. P.K., S.P., B.C.S.-S., P.S., and F.M.S. co-wrote the manuscript. W.A. and S.P. were responsible for AFLP wetlab. P.K. and E.Z. adapted and fine-tuned the RADseq protocol for all taxa and performed RADseq library preparation. W.A., P.K., B.C.S.-S., and F.M.S conceived analyses and figures. W.A., P.K. and S.P. performed all analyses. P.K. and S.P. prepared all figures. P.K., B.C.S.-S., P.S., F.M.S., E.Z., and members of the Steppe Consortium collected samples in the field. All authors contributed to the development of the manuscript and improved earlier drafts of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kirschner, P., Arthofer, W., Pfeifenberger, S. et al. Performance comparison of two reduced-representation based genome-wide marker-discovery strategies in a multi-taxon phylogeographic framework. Sci Rep 11, 3978 (2021). https://doi.org/10.1038/s41598-020-79778-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-79778-x
This article is cited by
-
Applying molecular and genetic methods to trees and their fungal communities
Applied Microbiology and Biotechnology (2023)
-
Recent contributions of molecular population genetic and phylogenetic studies to classic biological control of weeds
BioControl (2023)
-
Performance comparison of gel and capillary electrophoresis-based microsatellite genotyping strategies in a population research and kinship testing framework
BMC Research Notes (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
