
Abstract
MZB1 is an endoplasmic reticulum (ER)-resident protein that plays an important role in the humoral immune response by enhancing the interaction of the μ immunoglobulin (Ig) heavy chain with the chaperone GRP94 and by augmenting the secretion of IgM. Here, we show that MZB1 is also expressed in plasmacytoid dendritic cells (pDCs). Mzb1−/− pDCs have a defect in the secretion of interferon (IFN) α upon Toll-like receptor (TLR) 9 stimulation and a reduced ability to enhance B cell differentiation towards plasma cells. Mzb1−/− pDCs do not properly expand the ER upon TLR9 stimulation, which may be accounted for by an impaired activation of ATF6, a regulator of the unfolded protein response (UPR). Pharmacological inhibition of ATF6 cleavage in stimulated wild type pDCs mimics the diminished IFNα secretion by Mzb1−/− pDCs. Thus, MZB1 enables pDCs to secrete high amounts of IFNα by mitigating ER stress via the ATF6-mediated UPR.
Similar content being viewed by others


Introduction
Plasmacytoid dendritic cells (pDCs) were initially discovered in human lymph nodes in the 1950′s1. Their morphology proved to be controversial, with a lymphoid appearance that changes to a dendritic cell-like upon activation. The plasma cell-like appearance led to their nomenclature as “Plasmacytoid dendritic cells”2. The defining feature of pDCs is their ability to produce high levels of Type I Interferon (IFN) in response to viruses and viral components that are recognized by endosomal innate immune receptors such as the Toll-like receptors (TLR) 7 and TLR93,4. Upon stimulation of the TLRs with an agonist, 60% of the pDCs transcriptome is dedicated to the expression and secretion of IFNα5, whereby each cell is capable of producing a staggering 1–3 picograms within 24 h5,6. Due to their high IFN production, pDCs play an important role in controlling the initial stage of viral infections. Secreted IFNα enhances the activation of NK cells, B and T cells3. Moreover, pDCs have also been found to be a major source of IFNα in inflammatory autoimmune diseases such as Systemic Lupus Erythematosus (SLE), psoriasis and Scleroderma7,8,9. As highly secretory cells, pDCs and plasma cells are the only hematopoietic cell type that depend on an augmented protein-folding ability of the endoplasmic reticulum (ER) for their survival10,11.
Accumulation of unfolded proteins in the ER results in an ER stress that can be alleviated by the activation of the unfolded protein response (UPR), which restores cellular proteostasis12. In particular, secretory cells require an enhanced protein-folding capacity of the ER which involves the sensing of ER stress, an increased expression of protein folding chaperones and the expansion of the ER. The UPR consists of three pathways, the inositol-requiring transmembrane kinase/endonuclease (IRE1), the PKR-like ER protein kinase (PERK) and the activating transcription factor 6 (ATF6) pathways12,13,14. Upon accumulation of misfolded proteins, BiP dissociation leads to the activation of these three transmembrane proteins15. Activation of IRE1 generates an active isoform of the transcription factor XBP1 that can dimerize with the activated isoform of ATF6 which is generated by its translocation to the Golgi and proteolytic cleavage15,16,17,18,19. ATF6 plays a crucial role in increasing levels of the ER chaperones BiP and GRP94 to help cope with the increased folding demands. Moreover, ATF6 and XBP1 regulate ER expansion by increased lipid biosynthesis, whereby ER membrane expansion can alleviate ER stress independent of an accompanying increase in ER chaperone levels20,21. A fine balance is maintained between the different pathways, whereby IRE1 and ATF6 are the protective ones that are activated early upon ER stress and mediate protein folding22. Upon continued unmitigated stress and failure to restore protein homeostasis the PERK pathway plays a more prominent role, leads to the expression of CHOP, which is involved in ER associated protein degradation (ERAD)23,24.
The ER resident protein MZB1 (pERP1) is abundantly expressed in innate-like B cells, including Marginal zone (Mz) B cells of the spleen and peritoneal B1 B cells, as well as in antibody-secreting cells25,26,27. Deletion of Mzb1 resulted in a reduced humoral response with plasma cells secreting lower levels of IgM following in vivo stimulation of B cells28,29. Proteomic analysis of MZB1 interaction partners identified many proteins involved in the UPR, including the chaperones BiP, GRP94 (Hsp90B1), the calcium-binding proteins calnexin and calreticulin and the protein disulphide isomerase ERp57 and PDIA625. MZB1 acts as a co-chaperone of GRP94 that is needed for its interaction with various client proteins, including immunoglobulins and integrins28,29,30. Recent transcriptome analysis of all hematopoietic cell types showed that Mzb1 mRNA is also expressed in the non-B cell lineage cell the pDCs31. Here, we examine the function of MZB1 in pDCs and find that the deletion of Mzb1 results in reduced IFNα upon TLR9-mediated stimulation. We show that this defect involves an impaired activation of the ER stress sensor ATF6 and a diminished expansion of the ER upon cell stimulation by TLR9.
Results
MZB1 is expressed in plasmacytoid dendritic cells and regulates IFNα secretion
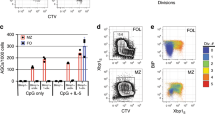
By immunoblot analysis of sorted splenic pDCs, we found that unstimulated pDCs express MZB1 (Fig. 1A). The level of MZB1 protein expression in pDCs was similar to that found in marginal zone B cells (MzB) and higher than that detected in follicular B cells (FoB). However, we could not determine whether MZB1 is uniformly expressed in pDCs because the anti-MZB1 antibody could not be used for intracellular flow cytometry. Treatment of wild type pDCs with a TLR9 agonist, CpG A oligonucleotide (ODN) that induces high IFNα production and only weakly stimulates NF-κB signaling32,33, did not change the level of MZB1 protein (Fig. 1B). To assess the function of MZB1 in pDCs, we first determined the numbers and frequencies of pDCs in Mzb1−/− mice. Flow cytometric analysis of Siglec H- and B220-double positive splenic pDCs indicated that their numbers and frequencies are similar in wild type and Mzb1 knockout mice (Supplementary Fig. S1A). Because the numbers of splenic pDCs are quite low, we expaned bone marrow-derived pDCs in culture with Flt3 ligand and sorted them by using the pDC markers Siglec H and B220 (Supplementary Fig. S1B). Stimulation of MZB1-deficient pDCs with CpG A resulted in an impaired IFNα secretion relative to the wild type pDCs, as determined by an Enzyme-Linked ImmunoSorbent Assay (ELISA) (Fig. 1C). In contrast, the secretion of IFNβ, which is expressed at a much lower level than that of IFNα, was unaffected by the deletion of Mzb1. To examine whether MZB1 is also required for the secretion of the pro-inflammatory cytokines IL6 and TNFα, we stimulated wild type and Mzb1−/− pDCs with CpG B, which activates TLR9 dependent NF-κB signaling32. The secretion of these cytokines, which were detected at a much lower level than that of IFNα, were not affected by the deletion of Mzb1 (Fig. 1D). Interestingly, stimulation Mzb1+/+ and Mzb1−/− pDCs with the TLR7 agonist Imiquimod, which is a weaker stimulator of IFNα secretion, resulted in a similar low level of IFNα secretion in wt and mutant cells (Fig. 1E). Taken together, these results suggest that MZB1 is required in conditions of very abundant secretion of IFNα.

MZB1 is expressed in pDCs and inactivation of Mzb1 affects IFNα secretion. (A) Immunoblot analysis of sorted follicular B (FoB) cells (CD19+, AA4.1-, CD23+ CD21+), Marginal Zone B (MzB) cells (CD19+, AA4.1-, CD23-, CD21+) and plasmacytoid dendritic cells (pDCs) (B220+, Siglec H+) to examine levels of MZB1 protein expression in each cell type. GAPDH is used as a loading control. (B) Immunoblot analysis of MZB1 protein level in splenic pDCs at various time points during a 24 h stimulation (image is representative of two experiments) with GAPDH used as a loading control. Uncropped blots are shown in Supplementary Fig. S3A, B. (C) ELISA-based quantification of secreted IFNα and IFNβ levels in the supernatants of 105 pDCs from Mzb1+/+ and Mzb1−/− mice at 24 h post stimulation with CpG A. Each dot represents an individual culture of sorted pDCs derived from the bone marrow of an individual mouse. (D) ELISA-based quantification of secreted TNFα and IL6 levels in the supernatant of 105 sorted pDCs at 24 h post stimulation with CpG B. (E) Secreted IFNα levels, quantified by ELISA, in the supernatant of 105 sorted Mzb1+/+ and Mzb1−/− pDCs at 24 h post stimulation with the TLR7 agonist Imiquimod. ns, non-significant. (F) Flow cytometry analysis of the surface expression of the activation marker CD86 on unstimulated and CpG A-stimulated pDCs. (G) Box plot of quantitative RT-PCR analysis to determine the relative mRNA levels of Ifna in Mzb1+/+ and Mzb1−/− pDCs at 6 and 24 h post stimulation with CpG A, normalized to 18S rRNA levels. The box borders represents the interquartile range and the horizontal line in the box is the median. Numbers of independent samples (n) are indicated below the graph. (H) qRT-PCR analysis of Tnfa (TNFα) and Il6 (IL6) transcript levels in Mzb1+/+ and Mzb1−/− pDCs stimulated with CpG A for 24 h. Transcript levels were normalized to those of 18S rRNA. Error bars show SD. Statistical difference between the mean was analysed by an unpaired two-tailed Student’s t-test. (***) P ≤ 0.0005. ns, non-significant.
MZB1-deficient pDCs have a normal TLR9 signaling response
MZB1 is a co-chaperone of GRP94, which is required for the proper expression and function of various TLRs28,34. Therefore, we examined whether the reduced IFNα secretion reflects a defect in TLR9 signaling. Stimulation of Mzb1+/+ and Mzb1−/− pDCs with CpG A resulted in similarly enhanced cell surface expression of the activation marker CD86, as determined by the flow cytometry analysis (Fig. 1F). Moreover, proper TLR9 signaling in Mzb1−/− pDCs was confirmed by the activation of integrin β1 (CD29), which was detected in flow cytometric analysis with an antibody that recognizes the extended conformation of CD2929 (Supplementary Fig. S1B). Finally, the stimulation of Mzb1+/+ and Mzb1−/− pDCs with CpG A resulted in similar increase in the levels of Ifna transcripts between 6hrs and 24 h after stimulation (Fig. 1G). As many different subtypes of IFNα exist, we used in this experiment degenerate primers that detect transcripts of multiple IFNα subtypes (Fig. 1G). However, we also confirmed the normal increase of Ifna transcripts in stimulated Mzb1−/− pDCs by using specific primers for transcripts of the IFNα1–2 subtypes (Supplementary Fig. S1C).
The marked up-regulation of IFNα expression involves a feed-forward loop in which IFNα augments its own expression by binding the IFN cell surface receptor (IFNAR) and activating the JAK-STAT pathway and the expression of IFN-stimulated genes35. Therefore, we also examined whether the observed IFNα secretion defect of stimulated Mzb1−/− pDCs is due to an impaired IFNα receptor signaling. A blockade of IFNAR signaling by an anti-IFNAR antibody has previously been shown to reduce IFNα secretion36. However, flow cytometric analysis to detect the surface expression of the IFNAR1 subunit did not reveal any significant differences between Mzb1+/+ and Mzb1−/− pDCs (Supplementary Fig. S1D). Moreover, the internalization of the IFNAR1, triggered by the binding of IFNα, was not affected by the deletion of Mzb1 (Supplementary Fig. S1E). The transcript levels of the IFNα-stimulated Tnfa and Il6 genes also increased to similar levels in wild type and MZB1-deficient pDCs upon stimulation with the CpG A (Fig. 1H). Taken together, these results show that the reduced secretion of IFNα in stimulated Mzb1−/− pDCs is neither due to a defective transcriptional response to TLR9 signaling nor to a defect in the IFNα feed-forward loop.
Mzb1 −/− pDCs show a reduced ER expansion and an intracellular retention of IFNα
During an unfolded protein response (UPR), the morphology of the ER undergoes an ATF6/XBP1-driven expansion of ER morphology20,21. ER dilation and changes in ER morphology have also been used to assess the ER stress status of cells37,38,39. To examine whether the impaired IFNα secretion in stimulated Mzb1−/− pDCs could reflect a reduced ER dilation, we evaluated the status of the ER by electron microscopy (EM) in control and stimulated conditions. The shape and morphology of the peripheral ER showed a similar structure of sheets in unstimulated Mzb1+/+ and Mzb1−/− pDCs (Fig. 2A). After 24 h of stimulation, the ER of Mzb1+/+ pDCs showed marked changes in morphology, whereas the Mzb1−/− pDCs maintained the elongated sheet structure (Fig. 2A). To quantify the changes in ER morphology, we calculated the sphericity indexes from the data of three independent experiments and determined that the sphericity of the ER in stimulated Mzb1−/− pDCs is approximately 30% lower than that of stimulated Mzb1+/+ pDCs (Fig. 2A).

Mzb1−/− pDCs show a reduced expansion of the ER and intracellular retention of IFNα. (A) Electron microscopy (EM) analysis of Mzb1+/+ and Mzb1−/− pDCs that were unstimulated or stimulated with CpG A for 24 h (left-hand panels). Red arrows indicate the ER. The quantification of the sphericity index of the ER, as the ratio of the width to the length of the ER, is shown in the right-hand panels. Each dot is a biologically separate sample with approximately 50 different cells imaged for each sample. Statistical difference between the mean was analysed by an unpaired two-tailed Student’s t-test. *P ≤ 0.05. Error bars show SD. ns, non-significant. (B) Intracellular and secreted IFNα levels of CpG A-stimulated (24hrs) Mzb1+/+ and Mzb1−/− pDCs, as determined by ELISA of lysates and supernatants of 105 cells, respectively. Each dot represents pDCs derived from the bone marrow of individual mice. Data from two separate experiments were included in the analysis. Statistical difference between the mean was analysed by an unpaired two-tailed Student’s t-test. *P ≤ 0.05, **P ≤ 0.005. Error bars show SD. (C) Co-immunoprecipitation assay to detect the interaction of MZB1 with IFNA2. Lysates of K46 B cells expressing HA-tagged IFNA2 were incubated with an anti-Mzb1 monoclonal antibody or an IgG control. MZB1 and IFNA2 were detected by immunoblot analysis with MZB1- and HA-specific antibodies, respectively. Uncropped blots are shown in Supplementary Fig. S3C.
The reduced sphericity index of stimulated Mzb1−/− suggests an impaired ER dilation and protein folding capacity. To examine whether the reduced IFNα secretion in Mzb1−/− pDCs reflects an accumulation of intracellular IFNα, we quantified the intracellular levels of IFNα by performing an ELISA with lysates from wild type and Mzb1−/− pDCs. A significantly higher level of intracellular IFNα was detected in Mzb1−/− pDC lysates as compared to Mzb1+/+ pDC lysates, suggesting that the Mzb1 deletion leads to a partial retention of IFNα in the ER (Fig. 2B). Conversely, the ELISA of the supernatants collected prior to cell lysis showed a reduced IFNα secretion by Mzb1−/− pDCs relative to that by Mzb1+/+ pDCs.
In antibody-secreting cells, MZB1 interacts with the immunoglobulin (Ig) μ heavy chain and enhances the secretion of IgM by acting as a co-chaperone of the substrate specific GRP94 chaperone26,27,28,29. Therefore, we examined whether MZB1 interacts with IFNα. As the anti-IFNα antibodies did not work in the immunoblot analysis, we examined an interaction in K46 B cells that had been transfected with an HA-tagged IFNA2 expression plasmid. We found that the HA-tagged IFNA2 is co-immunoprecipitated with an anti-MZB1 antibody (Fig. 2C). These results indicate that the ER-localized co-chaperone MZB1 interacts with IFNα and may enhance its proper folding in conditions of abundant secretion of IFNα.
Mzb1 −/− pDCs have a reduced ER stress response and impaired ATF6 activation
To further examine the molecular basis for the expansion of the ER and the impaired secretion of IFNα, we examined the UPR in wild type and Mzb1−/− pDCs. In response to ER stress, the ATF6 pathway has been implicated in the up-regulation of the Hspa5 (encoding BiP) during the early phase of the UPR12. Analysis of the RNA expression of Hspa5 by quantitative RT-PCR revealed a weak up-regulation in stimulated Mzb1−/− pDCs, relative to stimulated Mzb1+/+ pDCs (Fig. 3A). No difference in Hspa5 expression was observed in unstimulated Mzb1+/+ and Mzb1−/− pDCs (Fig. 3B). Thus, the diminished upregulation of BiP may lead to continued ER stress, activation of PERK pathway and increased expression of CHOP. Indeed, the level of Ddit3 (encoding CHOP) mRNA expression in Mzb1−/− pDCs was found to be higher than that in wild type pDCs, suggesting a problem in alleviating the ER stress by a proper UPR in the MZB1-deficient cells (Fig. 3A,B).

Mzb1−/− pDCs exhibit an improper ER stress response due to the impaired activation of the ATF6 pathway. (A) Box plot of qRT-PCR analysis to determine the relative Hspa5 (BiP) and Ddit3 (CHOP) mRNA levels after stimulation of Mzb1+/+ and Mzb1−/− pDCs with CpG A for 24 h. mRNA levels were normalized to those of 18S rRNA. Numbers of independent samples (n) are indicated below the graph. (B) qRT-PCR analysis of Hspa5 (BiP) and Ddit3 (CHOP) mRNA at basal levels in unstimulated pDCs. (C) Immunoblot analysis to detect uncleaved and cleaved ATF6 (approx. 55 kDa) in CpG A-stimulated pDCs. GAPDH serves as a loading control. IgH indicates residual IgH from the serum and * indicates an unknown cross-reacting protein. Uncropped blots are shown in Supplementary Fig. S3D. (D) ELISA-based quantification of the secreted IFNα in the supernatant of 105 Mzb1+/+ and Mzb1−/− pDCs that were untreated or treated with the ATF6 inhibitor AEBSF at 100 µM. (E) ELISA-based quantification of secreted IFNα in the supernatant of Mzb1+/+ and Mzb1−/− pDCs, untreated or treated with the ATF6 inhibitor Ceapin A7 at 3 µM. Each dot represents an individual pDC culture derived from an individual mouse. Statistical difference between the mean was analyzed by an unpaired two-tailed Student’s t-test. *P ≤ 0.05; **P ≤ 0.005. Error bars show SD. ns, non-significant.
Interestingly, the expanded ER morphology observed in stimulated Mzb1+/+ pDCs is reminiscent of the ER morphology seen in CHO cells that overexpress the cleaved and activated transcription factor ATF621. Therefore, we examined the possibility that MZB1 is involved in the activation of ATF6 under ER stress conditions. Immunoblot analysis indicated that stimulated Mzb1−/− pDCs have modestly reduced levels of full-length ATF6 but significantly lower levels of the ~ 55kD cleaved form of ATF6 relative to stimulated wild type pDCs (Fig. 3C). Moreover, we examined whether or not the pharmacological inhibition of ATF6 cleavage in stimulated wild type pDCs would result in an impaired IFNα secretion. To this end, we used the serine protease inhibitor AEBSF, which inhibits the first step of ATF6 cleavage in the Golgi40, and Ceapin A7, which blocks the transport of ATF6 to the Golgi41,42. Notably, the treatment of wild type pDCs with either inhibitor showed a similar reduction in IFNα secretion as observed in Mzb1−/− pDCs (Fig. 3D,E). Finally, we examined whether the MZB1 deficiency affects the IRE1 pathway and detected no obvious differences in the spliced Xbp1 levels between stimulated wild type and Mzb1−/− pDCs (Supplementary Fig. S1F). Taken together, our data suggest that Mzb1−/− pDCs have a reduced ability of activating the ATF6 pathway which may account for the reduced Hspa5 (BiP) expression and the impaired expansion of the ER.
Mzb1 −/− pDCs have an impaired ability to stimulate plasma cell differentiation
IFNα and pDCs play a role in enhancing humoral immunity by activating B and T cells43,44. To explore the functional consequences of the reduced IFNα secretion by Mzb1−/− pDCs on B cell activation, we performed co-culture experiments. MACS-sorted wild type CD19-positive B cells were co-cultured with either Mzb1+/+ or Mzb1−/− pDCs. The cultures were stimulated with CpG C and anti-IgM for 72 h (Fig. 4A). Flow cytometric analysis indicated that the percentage of CD138-positive plasmablasts is lower in the co-culture with Mzb1−/− pDCs as compared to the co-cultures with Mzb1+/+ pDCs (Fig. 4B,C). By performing ELISA with cell culture supernatants, we also detected lower levels of IgM in the co-cultures with the Mzb1−/− pDCs relative to the co-cultures with the wild type pDCs (Fig. 4D). Moreover, we observed a reduced surface expression of the CD86 activation marker on B cells that were co-cultured with Mzb1−/− pDCs (Fig. 4E).

Impaired B cell differentiation towards plasma cells upon co-culture with Mzb1−/− pDCs. (A) Schematic representation of co-cultures of MACS-sorted wild type CD19 B cells together with Mzb1+/+ or Mzb1−/− pDCs that were stimulated with both CpG C and anti-IgM for 72 h. (B) Flow cytometric analysis of the co-cultures 72 h post stimulation for plasmablast markers B220 and CD138 (Syndecan). (C) Quantification of percentage of CD138+ cells. (D) ELISA-based quantification of secreted IgM after co-culturing CD19+ B cells with pDCs for 72 h. (E) Quantification of the mean fluorescence intensity (MFI) of the B cell activation marker CD86 by flow cytometric analysis. (F) Flow cytometric analysis of plasma cell differentiation in co-cultures of CD19+ B cells with Mzb1+/+ pDCs in the absence or presence of IFNAR blocking antibody. (G) Quantification of the frequencies of plasmablasts in co-cultures of CD19+ B cells with Mzb1+/+ pDCs with or without the IFNAR blocking antibody (black) and in co-cultures of B cells and Mzb1−/− pDCs with or without exogenous IFN⍺ (red). (H) Flow cytometric analysis of plasma cell differentiation in co-cultures of B cells with Mzb1−/− pDCs in the presence or absence of exogenous IFN⍺. (I) Quantification of the MFI of the activation marker CD86 in B cells, co-cultured with Mzb1+/+ pDCs in the presence or absence of IFNAR blocking antibody (black) and co-cultured with Mzb1−/− pDCs with or without exogenous IFN⍺ (red). Each dot represents co-cultures with pDCs derived from an individual mouse. Statistical difference between the mean was analyzed by an unpaired two-tailed Student’s t-test. *P ≤ 0.05 **P ≤ 0.005. Error bars show SD.
To determine whether the impaired stimulation of plasma cell differentiation by Mzb1−/− pDCs is a result of the reduced IFNα secretion, we included an IFNAR blocking antibody in co-cultures of Mzb1+/+ pDCs to reduce the secreted IFNα levels. In this experiment, we also observed an impaired plasmablast differentiation (Fig. 4F,G). Conversely, the reduced differentiation of plasmablasts in co-cultures with Mzb1−/− pDCs could be rescued by the addition of exogenous IFNα (Fig. 4G,H). The effects of altered IFNα levels on plasmablast differentiation were also reflected by equivalent changes in the surface expression of CD86 (Fig. 4I). Thus, the reduced ability of IFNα secretion in Mzb1−/− pDCs results in an impaired stimulation of B cell differentiation towards antibody-secreting plasmablasts.
Discussion
pDCs are well known for their capacity to secrete high levels of IFNα after TLR9 stimulation during viral infections. High levels of protein secretion lead to increased ER stress in these cells and result in the activation of the unfolded protein response (UPR). We and others have previously described the role of MZB1 as an ER resident co-chaperone of GRP94 that enhances the secretion of IgM and IgA and promotes plasma cell differentiation25,26,28,45. Here we show that MZB1 is expressed in pDCs and is required for abundant IFNα secretion, which is a major hallmark of pDCs. MZB1 is a member of a small family of CNPY co-chaperones that interact with GRP9434,46. In particular, CNPY3, which is ubiquitously expressed and localized in the ER, acts a GRP94 co-chaperone that enhances the folding of TLR434. Therefore, we considered the possibility that MZB1 affects the function of TLR7 and/or TLR9. In MZB1-deficient pDCs, however, we observed a normal TLR9 signaling response, as evidenced by the similar transcriptional activation of the Ifna genes in mutant and wild type pDCs. Moreover, in Mzb1−/− pDCs we detected higher amounts of intracellular IFNα as compared to Mzb1+/+ pDCs, suggesting that IFNα is produced but not properly secreted, possibly due to an impaired protein folding in the ERIn antibody-secreting B cells, MZB1 has been shown to augment the interaction of the Ig μ heavy chain with the chaperone GRP94 in conditions of abundant Ig expression28, and therefore, the observed interaction of MZB1 with IFNα suggests a similar mechanism of MZB1 function in pDCs. According to this view, the interaction of MZB1 with IFNα occurs in the ER and enhances the folding of IFNα in conditions of abundant protein expression and ER stress. In contrast, the function of MZB1 is not required for the weak IFNα secretion in Imiquimod-stimulated pDCs, in which the expression of IFNα is only modestly enhanced relative to unstimulated pDCs.
Inefficient protein folding can be the result of an impaired ER stress response. Indeed, we observed two hallmarks of unresolved ER stress. First, we found that Mzb1−/− pDCs do not upregulate the major protein folding chaperone BiP. In addition, the EM analysis of stimulated pDCs indicated that Mzb1−/− pDCs do not acquire the dilated ER morphology detected in Mzb1+/+ pDCs. Both features of ER stress have been linked to the ATF6 pathway of the UPR12,21,47. In particular, the activation of ATF6 involves the dissociation of BiP, translocation to the Golgi, proteolytic cleavage and subsequent nuclear translocation is responsible for the transcriptional upregulation of the chaperone genes16,18,19. Moreover, changes in the abundance of activated ATF6 by overexpression have been linked to changes in ER morphology21. Therefore, we propose a model in which MZB1 enhances the interaction of BiP with the client IFNα, resulting in a default liberation of ATF6 from BiP as an initial step in its activation (Supplementary Fig. S2A,B). CNPY2, a ubiquitously expressed member of the CNPY family of co-chaperones, has been shown to initiate the PERK pathway of the UPR48. In contrast, MZB1 is selectively expressed in cells with a high secretory demand, which may enable these cells to reach their full secretory capacity. Indeed, all known roles for MZB1 have been linked to the efficient secretion and protein folding in conditions of ER stress.
pDCs have been known to play a role in linking innate and adaptive immunity. Type I interferons have been shown to enhance humoral immunity and promote immunoglobulin isotype switching49. Moreover, CpG-stimulated pDCs induce plasma cell differentiation in naïve B cells50. Indeed, we find that the differentiation of B cells towards plasma cells is augmented by the co-culturing of B cells with stimulated wild type pDCs. In Mzb1−/− pDCs, however, the reduced IFNα secretion resulted in diminished differentiation towards plasma cells and in lower levels of IgM secretion. The function of MZB1 in pDCs could be relevant during chronic viral and microbial infections in which the IFNα secretion enables an innate immune response and stimulates B cells to differentiate into antibody-secreting plasma cells. Furthermore, IFNα plays a crucial role in autoimmune diseases like psoriasis and SLE3,8. In murine psoriasis xenograft models in which pDCs infiltrate the skin, the inhibition of IFNα secretion has been found to ameliorate the disease symptoms. In SLE, MZB1 expression is frequently upregulated51, which could account for the enhanced secretion of IFNα and the augmented secretion of auto-antibodies by B cells. The increased IFNα levels in SLE patients as a driver of disease progression also correlate with the reduced frequency of immunosuppressive regulatory B cells (Breg)52. Clinical studies have shown that blocking type I interferons helps to alleviate SLE symptoms and disease progression. MZB1 has a more restricted expression pattern than IFNα, which makes it an attractive potential therapeutic target in autoimmune diseases.
Materials and methods
Cell culture and cell isolation
Bone marrow cells from mice were prepared following the standard protocol. Cells were counted using the Casy Cell Counter and suspended at 2 × 106 cells/ml in RPMI with 10% FCS, Penicillin, Streptomycin, 2 mM L-Glutamine and beta-mercaptoethanol, supplemented with 200 ng/ml Flt3 ligand. On day 8, fully developed pDCs were sorted by flow cytometry for the surface expression of both B220 and Siglec H. Cells of the K46 B cell line were transfected with an Ifna2-pMys-expression plasmid in which an HA-tag is attached to the C-terminus of IFNa2. Splenic pDCs were MACS-sorted with a mouse Plasmacytoid dendritic cell isolation kit (Miltenyi Biotech) and subsequently sorted by flow cytometry FACS.
Cloning and retroviral transduction
For the expression of HA-tagged IFNA2 protein, Ifna2 cDNA was cloned with an optimized Kozak sequence at the 5′ end and HA-tag sequence at the 3′end into a pMYs-IRES-GFP bicistronic vector to allow for the expression of IFNA2 along with GFP. For the production of HA-IFNA2-expressing retroviruses, platE cells were transfected with the expression plasmid. Supernatant of transfected Plat E cell was used for transduction of K46 B cells and GFP-expressing cells were sorted prior to the preparation of cell lysates for co-immunoprecipitation.
CpG oligonucleotide-mediated stimulation, inhibition of ATF6 cleavage and B cell co-cultures
Sorted bone marrow-pDCs were aliquoted into 96 well plates at 100,000 cells per well. Cells were stimulated with 1 mM of CpG A (ODN1585) or B (ODN1886) for 24 h, unless otherwise stated. For the pharmacological inhibition of the ATF6 pathway, cells were treated with AEBSF at a final concentration of 100 µM or with Ceapin A7 (Sigma) at a concentration of 3 µM, four hours post stimulation with CpG ODN. For co-culture experiments 100,000 CD19-positive wild type B cells, purified by a positive MACs selection, were co-cultured with 30,000 FACS-sorted bone marrow-derived pDCs and stimulated with 1 mM CpG C (ODN M362) and anti-IgM for 72 h as previously described in50. All ODNs used were obtained from Invivogen.
RNA isolation and RT-PCR
RNA was isolated following homogenisation in Trizol according to the manufacturer’s instructions. cDNA was reverse transcribed using Thermo Fischer cDNA kit. RT-PCR analysis was carried out using Sybr green reagents. Primers used are listed in the Supplemental Information.
Cytokine quantification by enzyme-linked immunosorbent assay (ELISA)
Measurements of the cytokines IFNα, IFNβ, IL6 and TNFα were performed by ELISA. The following kits were used according to the manufacturer’s protocol. Mouse IFN-alpha Platinum ELISA-4, Mouse IL-6 Ready-SET-go, Mouse TNF-alpha Ready-Set-go (eBiosciences) and LegendMax Mouse IFNβ ELISA kit (BioLegend).
Immunoprecipitations and immunoblot analysis
Cells were resuspended in 20 mM HEPES pH7.6, 1 × Protease inhibitor mix, PMSF, 0.1% NP40, 10 mM Sodium orthovanadate, 2 mM magnesium chloride, 150 mM Sodium chloride and 10% glycerol, followed by three sonications for 30 secs. The protein concentrations were measured by Bradford assays, and the samples were diluted with Laemmli buffer and run on 4–12% SDS PAGE gels. The proteins were transferred onto a membrane and probed with the appropriate antibodies.
Immunoprecipitations of MZB1 were carried out by using Protein-G Dyna-beads which were blocked overnight with 1% BSA. Following a 4-h immunoprecipitation at 4 °C, the beads were washed with the lysis buffer five times. After gel electrophoresis, the samples were visualized by immunoblot analysis with antibodies listed in the Supplemental Information. Co-immunoprecipitation experiments to examine the interaction between MZB1 and HA-tagged INFA2 in transfected K46 B cells were performed by treating cells with a BD Golgi stop (4 µl/6 ml culture medium) for 5 h and then lysed in the buffer described above.
FACS analysis
Single-cell suspensions were resuspended in PBS with 2% FCS and stained for flow cytometric analysis. Antibodies used are listed in the Supplemental Information. Data were acquired with an LSR Fortessa (BD Biosciences) and analyzed using the Flow jo software (version 10.1r5; https://www.flowjo.com/solutions/flowjo/downloads/previous-versions).
Electron microscopy
Cells were pelleted and resuspended in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer pH 7.4 and incubated for 20 min at RT. Subsequently, reduced IFNα secretion capacity he cells were pelleted and resuspended in 0.1 M sodium cacodylate pH7.4 and stored at 4 °C for further processing. After dehydration, the samples were embedded in Eponate 12 resin (Ted Pella) and sections were cut. Images were acquired using a FEI Tecnai12 Transmission electron microscope equipped with a TIETZ digital camera. ER enlargement was analyzed by sphericity index. Briefly, major and minor axis were measured for each ER compartment identified in 50 randomly selected images per condition. Sphericity index was calculated according to the formula major/minor axis.
Mouse lines and in vivo experiments
All mouse experiments were carried out in accordance to the guidelines of the Federation of European Laboratory Animal Science Association and following legal approval of the animal committee of the government bureau (Regierungspräsidium) Freiburg, Germany (license Gr-iTO-1). Mzb1+/+ and Mzb1−/− mice were generated as previously described28. Mouse strains were bred and maintained in the animal care facility of the Max Planck Institute of Immunobiology and Epigenetics. Experiments were performed with 7 to 14-week-old mice in backcrossed SV129 background.
References
Lennert, K. & Remmele, W. Karyometric research on lymph node cells in man. I. Germinoblasts, lymphoblasts & lymphocytes. Acta Haematol. 19, 99–113 (1958).
McKenna, K., Beignon, A. S. & Bhardwaj, N. Plasmacytoid dendritic cells: Linking innate and adaptive immunity. J. Virol. 79, 17–27. https://doi.org/10.1128/JVI.79.1.17-27.2005 (2005).
Gilliet, M., Cao, W. & Liu, Y. J. Plasmacytoid dendritic cells: Sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 8, 594–606. https://doi.org/10.1038/nri2358 (2008).
Cella, M. et al. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat. Med. 5, 919–923. https://doi.org/10.1038/11360 (1999).
Ito, T., Kanzler, H., Duramad, O., Cao, W. & Liu, Y. J. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood 107, 2423–2431. https://doi.org/10.1182/blood-2005-07-2709 (2006).
Siegal, F. P. et al. The nature of the principal type 1 interferon-producing cells in human blood. Science 284, 1835–1837 (1999).
Eloranta, M. L. & Ronnblom, L. Cause and consequences of the activated type I interferon system in SLE. J. Mol. Med. (Berl) https://doi.org/10.1007/s00109-016-1421-4 (2016).
Nestle, F. O. et al. Plasmacytoid predendritic cells initiate psoriasis through interferon-alpha production. J. Exp. Med. 202, 135–143. https://doi.org/10.1084/jem.20050500 (2005).
Swiecki, M. & Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 15, 471–485. https://doi.org/10.1038/nri3865 (2015).
Dhodapkar, M. V. Protein stress? pDCs go Toll-free. Blood 117, 376–377. https://doi.org/10.1182/blood-2010-11-315838 (2011).
Iwakoshi, N. N., Pypaert, M. & Glimcher, L. H. The transcription factor XBP-1 is essential for the development and survival of dendritic cells. J. Exp. Med. 204, 2267–2275. https://doi.org/10.1084/jem.2007052.5 (2007).
Walter, P. & Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 334, 1081–1086. https://doi.org/10.1126/science.1209038 (2011).
Ron, D. & Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell. Biol. 8, 519–529. https://doi.org/10.1038/nrm2199 (2007).
Sitia, R. & Braakman, I. Quality control in the endoplasmic reticulum protein factory. Nature 426, 891–894. https://doi.org/10.1038/nature02262 (2003).
Bertolotti, A., Zhang, Y., Hendershot, L. M., Harding, H. P. & Ron, D. Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat. Cell. Biol. 2, 326–332. https://doi.org/10.1038/35014014 (2000).
Yamamoto, K. et al. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell. 13, 365–376. https://doi.org/10.1016/j.devcel.2007.07.018 (2007).
Nadanaka, S., Okada, T., Yoshida, H. & Mori, K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol. Cell. Biol. 27, 1027–1043. https://doi.org/10.1128/MCB.00408-06 (2007).
Haze, K., Yoshida, H., Yanagi, H., Yura, T. & Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell. 10, 3787–3799 (1999).
Shen, J., Chen, X., Hendershot, L. & Prywes, R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell. 3, 99–111 (2002).
Schuck, S., Prinz, W. A., Thorn, K. S., Voss, C. & Walter, P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell. Biol. 187, 525–536. https://doi.org/10.1083/jcb.200907074 (2009).
Bommiasamy, H. et al. ATF6alpha induces XBP1-independent expansion of the endoplasmic reticulum. J. Cell. Sci. 122, 1626–1636. https://doi.org/10.1242/jcs.045625 (2009).
Todd, D. J., Lee, A. H. & Glimcher, L. H. The endoplasmic reticulum stress response in immunity and autoimmunity. Nat. Rev. Immunol. 8, 663–674. https://doi.org/10.1038/nri2359 (2008).
Oyadomari, S. & Mori, M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell. Death Differ. 11, 381–389. https://doi.org/10.1038/sj.cdd.4401373 (2004).
Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell. Biol. 13, 89–102. https://doi.org/10.1038/nrm3270 (2012).
Flach, H. et al. Mzb1 protein regulates calcium homeostasis, antibody secretion, and integrin activation in innate-like B cells. Immunity 33, 723–735. https://doi.org/10.1016/j.immuni.2010.11.013 (2010).
van Anken, E. et al. Efficient IgM assembly and secretion require the plasma cell induced endoplasmic reticulum protein pERp1. Proc. Natl. Acad. Sci. U.S.A. 106, 17019–17024. https://doi.org/10.1073/pnas.0903036106 (2009).
Shimizu, Y., Meunier, L. & Hendershot, L. M. pERp1 is significantly up-regulated during plasma cell differentiation and contributes to the oxidative folding of immunoglobulin. Proc. Natl. Acad. Sci. U.S.A. 106, 17013–17018. https://doi.org/10.1073/pnas.0811591106 (2009).
Rosenbaum, M. et al. MZB1 is a GRP94 cochaperone that enables proper immunoglobulin heavy chain biosynthesis upon ER stress. Genes Dev. 28, 1165–1178. https://doi.org/10.1101/gad.240762.114 (2014).
Andreani, V. et al. Cochaperone Mzb1 is a key effector of Blimp1 in plasma cell differentiation and beta1-integrin function. Proc. Natl. Acad. Sci. U.S.A. 115, E9630–E9639. https://doi.org/10.1073/pnas.1809739115 (2018).
Eletto, D., Dersh, D. & Argon, Y. GRP94 in ER quality control and stress responses. Semin. Cell. Dev. Biol. 21, 479–485. https://doi.org/10.1016/j.semcdb.2010.03.004 (2010).
Heng, T. S., Painter, M. W. & Immunological Genome Project C. The immunological genome project: Networks of gene expression in immune cells. Nat. Immunol. 9, 1091–1094. https://doi.org/10.1038/ni1008-1091 (2008).
Vollmer, J. et al. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur. J. Immunol. 34, 251–262. https://doi.org/10.1002/eji.200324032 (2004).
Jeske, S., Pries, R. & Wollenberg, B. CpG-Induced IFN-alpha production of plasmacytoid dendritic cells: Time and dosage dependence and the effect of structural modifications to the CpG backbone. Nucleic Acid Ther. 23, 118–124. https://doi.org/10.1089/nat.2012.0384 (2013).
Liu, B. et al. Folding of Toll-like receptors by the HSP90 paralogue gp96 requires a substrate-specific cochaperone. Nat. Commun. 1, 79. https://doi.org/10.1038/ncomms1070 (2010).
Platanias, L. C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 5, 375–386. https://doi.org/10.1038/nri1604 (2005).
Wilson, E. B. et al. Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340, 202–207. https://doi.org/10.1126/science.1235208 (2013).
Akiyama, M. et al. Increased insulin demand promotes while pioglitazone prevents pancreatic beta cell apoptosis in Wfs1 knockout mice. Diabetologia 52, 653–663. https://doi.org/10.1007/s00125-009-1270-6 (2009).
Riggs, A. C. et al. Mice conditionally lacking the Wolfram gene in pancreatic islet beta cells exhibit diabetes as a result of enhanced endoplasmic reticulum stress and apoptosis. Diabetologia 48, 2313–2321. https://doi.org/10.1007/s00125-005-1947-4 (2005).
Oslowski, C. M. & Urano, F. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol. 490, 71–92. https://doi.org/10.1016/B978-0-12-385114-7.00004-0 (2011).
Okada, T. et al. A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J. Biol. Chem. 278, 31024–31032. https://doi.org/10.1074/jbc.M300923200 (2003).
Gallagher, C. M. et al. Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6alpha branch. Elife https://doi.org/10.7554/eLife.11878 (2016).
Gallagher, C. M. & Walter, P. Ceapins inhibit ATF6alpha signaling by selectively preventing transport of ATF6alpha to the Golgi apparatus during ER stress. Elife https://doi.org/10.7554/eLife.11880 (2016).
Jego, G. et al. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 19, 225–234 (2003).
Fonteneau, J. F. et al. Activation of influenza virus-specific CD4+ and CD8+ T cells: A new role for plasmacytoid dendritic cells in adaptive immunity. Blood 101, 3520–3526. https://doi.org/10.1182/blood-2002-10-3063 (2003).
Xiong, E. et al. MZB1 promotes the secretion of J-chain-containing dimeric IgA and is critical for the suppression of gut inflammation. Proc. Natl. Acad. Sci. U.S.A. 116, 13480–13489. https://doi.org/10.1073/pnas.1904204116 (2019).
Anelli, T. & van Anken, E. Missing links in antibody assembly control. Int. J. Cell. Biol. 2013, 606703. https://doi.org/10.1155/2013/606703 (2013).
Okada, T., Yoshida, H., Akazawa, R., Negishi, M. & Mori, K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem. J. 366, 585–594. https://doi.org/10.1042/BJ20020391 (2002).
Hong, F. et al. CNPY2 is a key initiator of the PERK-CHOP pathway of the unfolded protein response. Nat. Struct. Mol. Biol. 24, 834–839. https://doi.org/10.1038/nsmb.3458 (2017).
Le Bon, A. et al. Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 14, 461–470 (2001).
Poeck, H. et al. Plasmacytoid dendritic cells, antigen, and CpG-C license human B cells for plasma cell differentiation and immunoglobulin production in the absence of T-cell help. Blood 103, 3058–3064. https://doi.org/10.1182/blood-2003-08-2972 (2004).
Hung, T. et al. The Ro60 autoantigen binds endogenous retroelements and regulates inflammatory gene expression. Science 350, 455–459. https://doi.org/10.1126/science.aac7442 (2015).
Menon, M., Blair, P. A., Isenberg, D. A. & Mauri, C. A regulatory feedback between plasmacytoid dendritic cells and regulatory B cells is aberrant in systemic lupus erythematosus. Immunity 44, 683–697. https://doi.org/10.1016/j.immuni.2016.02.012 (2016).
Acknowledgements
We are grateful to Drs. Eirini Trompouki and Marta Derecka for critical reading of the manuscript and Marika Rott for the assistance with the manuscript preparation. We are thankful to Drs. David Sanin and Katarzyna Grzes for fruitful discussions. We thank Dr. Pierre Cauchy for help with the statistical analysis, Ingrid Falk for technical assistance and members of the Grosschedl lab for discussions. We thank the FACS and animal facilities of the MPI. This work was supported by funds from the Max Planck Society.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
T.K. conceived the study and performed the experiments. M.C. and E.L.P.. analysed EM data. E.J.P. was involved in critical discussions and sharing of protocols. R.G. conceived and supervised the study. T.K. and R.G. wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kapoor, T., Corrado, M., Pearce, E.L. et al. MZB1 enables efficient interferon α secretion in stimulated plasmacytoid dendritic cells. Sci Rep 10, 21626 (2020). https://doi.org/10.1038/s41598-020-78293-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-78293-3
This article is cited by
-
Proteomic analysis identifies subgroups of patients with active systemic lupus erythematosus
Clinical Proteomics (2023)
-
Comparison of immune cells and diagnostic markers between spondyloarthritis and rheumatoid arthritis by bioinformatics analysis
Journal of Translational Medicine (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
