
Abstract
Using environmental DNA (eDNA) metabarcoding, we compared fish diversity in two distinct water bodies within the Yangtze River Basin with known populations of the critically endangered Yangtze finless porpoise (Neophocaena asiaeorientalis; YFP): the Tian-e-Zhou Reserve and Poyang Lake. We aimed to create a fish surveying tool for use in the Yangtze River Basin, while also gaining a better understanding of the prey distribution and diversity within two of the remaining strongholds of YFP. 16S rRNA universal primers were developed to amplify fish eDNA. After high-throughput sequencing and stringent data filtering, we identified a total of 75 fish species (6 orders, 9 families, 57 genera) across seasons and regions. Nine of the 75 fish species were among the 28 known YFP prey species, three of which were detected in all water samples. Our eDNA metabarcoding identified many species that had been previously captured using traditional netting practices, but also numerous species not previously collected in these water bodies. Fish diversity was higher in Poyang Lake than in Tian-e-Zhou Reserve, as well as higher in the spring than in summer. These methods provide a broadly applicable tool to quantify fish diversity and distributions throughout the Yangtze River Basin, and to inform conservation strategies of YFP.
Similar content being viewed by others


Introduction
Globally, aquatic environments and biodiversity face myriad human-caused threats including pollution, invasive species, overharvesting, diversion, drainage, and climate change1. In particular, the 2018 World Wildlife Fund Living Planet Index suggests that freshwater species numbers have declined by 83% on average since 1970, a rate faster than either terrestrial or marine species2. Our ability to assess the degree of threat or vulnerability of any species requires that we can reliably detect, quantify, and monitor populations in the wild. Indeed, the conservation of any species requires that we have good data on population demography, distribution and migration patterns, and species ecology. One major cause for the decline of aquatic species of conservation concern is the diminution of prey abundance or changes in their phenology or distribution1, precluding the long-term population persistence for many threatened predatory species.
The Yangtze finless porpoise (Neophocaena asiaeorientalis; YFP) was once considered a subspecies of the narrow-ridged finless porpoise, but was recognized as a separate species in 20183,4. It inhabits the middle and lower reaches of the Yangtze River, as well as larger tributaries or lakes connected to the river, notably Poyang Lake and Dongting Lake, and is the only freshwater porpoise species in the world4. The Yangtze finless porpoise has been listed as Critically Endangered by the IUCN Red List since 20135. The decrease in abundance and diversity of prey of the YFP, particularly fish, is suggested to be a major contributing factor to its decline6.
First established in 1992 by the Chinese government for the Yangtze River Dolphin, Lipotes vexillifer7, the Tian-e-Zhou Baiji National Natural Reserve (hereafter Tian-e-Zhou Reserve) located in Shishou City, Hubei Province, China, is an oxbow of the Yangtze River. It is the main sanctuary for the YFP8 containing more than 60 individuals (as of 2015)9. The reserve is 21 km long, up to 1–2 km wide, and contains three functional areas: core, buffer and test zones; porpoises are most frequently observed in the core zone, followed by the buffer zone10. Importantly, YFP individuals within the Tian-e-Zhou Reserve reproduce and the population increases annually, seeming to thrive without food supplementation7. Poyang Lake contains almost half of the extant YFP population (approximately 450 individuals)11, but little is known about their feeding habits. Tools to assess the distribution and diversity of prey species would thus provide important insights into the management and recovery of this critically endangered cetacean.
Environmental DNA (eDNA) metabarcoding is a noninvasive and powerful approach for surveying biodiversity and has been applied to numerous taxa including fish12,13,14,15, other vertebrates13,16,17, invertebrates18 and zooplankton19. It requires only that we collect environmental samples (e.g. water, soil, air or snow), then extract the total DNA contained therein20. Environmental DNA is released into environments via species secretions, excretion, shedding, gametes or carcasses21,22,23. After collecting environmental samples and extracting the DNA, researchers can target single gene regions across multiple taxa using conserved primers and obtain data on their presence and relative abundance in a single sample24.
Here we apply eDNA metabarcoding to quantify fish diversity in the Tian-e-Zhou Reserve and Poyang Lake, with the goals of (i) creating and testing a tool to survey a broad array of fish diversity in the Yangtze River Basin, and (ii) understanding prey distribution and diversity within two of the remaining strongholds of YFP. By investigating these patterns, we may gain insights on fish communities and seasonal and spatial changes in species richness and relative abundances within the Yangtze River Basin. Quantifying variability in YFP prey fish species will also provide an important tool for managing the habitats of this critically endangered cetacean.
Materials and methods
eDNA sampling
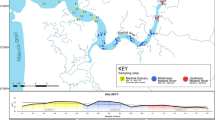
We collected water samples in spring (April 12 to April 18, 2017) and summer (July 3 to July 6, 2017, and July 23 to July 26, 2017), sampling 5 sites in the Tian-e-Zhou Reserve (A through E), and 10 sites in Poyang Lake (a through j) (Fig. 1). Sampling sites spanning the entire length of the Tian-e-Zhou Reserve were identical to those in Stewart et al.10. Sites A, B, C and D were located in the core zone (main YFP inhabited area), and site E in the buffer zone (less inhabited area). In Poyang Lake, sampling sites were selected to coincide with locations where local fishermen visually observed YFP frequently. Geographical coordinates for each sampling site were recorded using a handheld GPS unit, and pH, temperature, dissolved oxygen (DO) concentration were measured using portable meters (Supplementary Table S1).

Geographical distributions of 15 sampling sites across 2 regions (insets): Tian-e-Zhou Baiji National Nature Reserve and Poyang Lake. Map created using QGIS version 3.2.0 (QGIS Development Team. 2018. QGIS Geographic Information System. Open Source Geospatial Foundation Project. Retrieved from https://qgis.osgeo.org).
At each sampling site in spring and summer, we used a VanDorn water sampler (GRASP CG-00, GRASP Science and Technology, Beijing, China) to collect 15, 1-L water samples 1 m from the bottom to avoid sediment, with a maximum collection depth of 13 m (Supplementary Table S1). Following standardized protocols to minimize the probability of cross contamination25,26 with minor modifications, the sampler was sterilized with 10% diluted bleach between all sampling events, and then rinsed three times with surface water at each site before each sample collection, ensuring that rinse site and actual sampling locale were at least 2–3 m apart. Water samples were filtered using a portable peristaltic pump (Spectra Field-Pro Professional Grade, Spectra Scientific, Toronto, Canada) with 47-mm diameter mixed cellulose-ester filter paper, 0.45 µm pore size27. The filter holder (Sterifil Filter Holder, MILLIPORE, Shanghai, China) was also sterilized with bleach and surface water between uses as described above. We used one or two filter papers to filter each 1-L water sample depending on turbidity and clarity of the water. We used 95% ethanol and flame-sterilized tweezers to fold and place the filter paper from filtering each 1-L water sample into a 5 mL Eppendorf tube filled with 95% ethanol. At each sampling site across the two seasons, we also collected a sampling negative control (1 L of distilled water per site that was treated identically to the above water filtering protocol) to test for contamination during the sampling process. All samples were stored in the lab at − 20 °C until eDNA extraction.
eDNA extraction
Following Thomsen et al.28 with minor adjustments to the protocol, each filter was air-dried, rolled up, cut into ca. 1 mm slices and placed in a 2.0 mL lysis tube. 0.3 g of 0.9–2.0 mm stainless steel beads and 740 μL of tissue lysis buffer ATL (QIAGEN) were added to each tube, which were then shaken in an Air Cooling Bullet Blender (Storm BBY24M, NEXT ADVANCE) at high-speed for 3 min. After this, the tubes were incubated at 56 °C for 30 min, followed by another shaking step and incubation step as above. Then 80 µL of 20 mg/mL Proteinase K Solution (BIOLINE) was added to each tube followed by a final incubation at 56 °C for 2 h with agitation. Tubes were then vortexed for 15 s and spun for 5 min (17,000 g). Each sample supernatant (500 μL) was transferred into a new 1.5 mL tube and spun for 1 min (17,000 g).
A volume of 500 µL of chloroform-isoamyl alcohol (24:1)29,30,31 was added to the supernatant of each sample. Each tube was vortexed briefly, shaken for 10 min on a shaker, and spun for 20 min (13,000 rpm). The supernatant (aqueous phase) was then transferred to a new 1.5 mL tube, and this chloroform-isoamyl alcohol step was repeated. 500 µL of isopropanol and 250 µL of 5 M NaCl solution were then added to each tube, which were then vortexed briefly, spun for 1 min (17,000 g), and incubated at − 20 °C overnight. Each tube was then spun for 15 min (15,000 g), and all supernatants were carefully poured off without losing the DNA pellet. 150 µL of 70% ethanol were added to further clean the DNA, with each tube spun for 15 min (15,000 g), and the ethanol carefully decanted. This step was repeated twice. After the residual ethanol was allowed to evaporate, DNA was resuspended by adding 100 µL of elution buffer AE (QIAGEN). Tubes were incubated at 56 °C for 5–10 min, vortexed briefly, and spun for 1 min (15,000 g).
For each sampling site across seasons, three 100 µL DNA extracts from 3 individual 1-L water samples were combined for a final volume of 300 µL (one eDNA sample). All eDNA samples and extracts from sampling negative controls were stored at − 20 °C until PCR. To avoid contamination, we carried out all eDNA extractions in a biosafety cabinet that was irradiated with UV for 30 min prior to processing. All plasticware and pipettes were also UV sterilized between extractions. Before each use, the lab bench was cleaned with freshly-prepared 2% diluted bleach solution.
Focal prey species
We searched the China Knowledge Network (www.cnki.net) using the term “Yangtze finless porpoise” (in Chinese 江豚) on December 14, 2017 to find articles related to the diet of the finless porpoise. Our final list included 490 papers. From these, we determined that the diet of the YFP was mainly composed of 14 fish species throughout the year (Supplementary Table S2). According to Yang et al.6, YFP diet may include an additional 14 fish species (Supplementary Table S2). For development of DNA primers, we thus targeted these 28 fish species, belonging to 4 orders, 8 families and 22 genera.
Primer design and initial PCR
We searched the NCBI (National Center for Biotechnology Information; ncbi.nlm.nih.gov) nucleotide database for complete mitochondrial genome sequences of these 28 YFP prey target fish species (Supplementary Table S2). We found a total of 198 16S rRNA sequences that we downloaded and then aligned using ClustalW32. We designed a pair of degenerate primers (forward primer, 16S_eDNAF 5′-GTACCTTTTGCATCATGATTYAG-3′ and reverse primer, 16S_eDNAR 5′-TCTYCCCACTCTTTTGC-3′) to amplify a short fragment approximately 133–140 bp in length (Fig. 2) using the BioEdit Sequence Alignment Editor33.

Schematic representation of the (a) PCR amplification and (b and c) paired-end library preparation using a two-step tailed PCR.
We initially tested this primer pair using DNA extracted from tissue samples of 9 fish species (Supplementary Table S2). These fish species were sampled from Poyang Lake in March 2017 using electrofishing licensed by the Duchang Fishery Bureau. All sampling was conducted in accordance with the Regulations of the People’s Republic of China for the Implementation of Wild Aquatic Animal Protection (1993), adhering to all ethical guidelines and legal requirements in China. We also made separate synthetic DNA solutions (Gene Universal, Delaware USA) of prey target sequences for the remaining 19 fish species for which we lacked tissue samples to validate primer specificity (for sequences see Supplementary Table S3). Fragments of the 16S rRNA gene were amplified in 25 μL PCR cocktails containing 12.5 μL of 2 × Taq Mix (including 0.25U/μL Taq DNA polymerase, 2X PCR buffer, 0.4 mM dNTPs, 3.2 mM MgCl2 and 0.02% bromophenol blue; Froggabio), 1.2 μL of 10 μM each primer, 0.5 μL of 10 mg/mL Bovine Serum Albumin (BSA; Thermo Fisher Scientific), 3–5 ng of template DNA and nuclease-free water. The PCR cycling conditions were as follows: an initial denaturation step at 95 °C for 5 min followed by 36 cycles of 96 °C for 30 s, 52 °C for 45 s, 72 °C for 45 s and a final extension at 72 °C for 7 min. We performed all PCRs in triplicate. The PCR products were visualized on 1.5% agarose gels stained with RedSafe™ Nucleic Acid Staining Solution. Size standards (100-bp ladder) were included in each gel to verify fragment sizes. Sanger sequencing results (The Centre for Applied Genomics, Toronto, Canada) of amplicons from PCR of DNA extracted from tissue samples and amplicons from PCR of every synthetic DNA solution confirmed the applicability of this primer pair.
For eDNA samples and extracts from field sampling negative controls, we followed the same PCR procedures described above, with the exception that DNA template and nuclease-free water volumes were adjusted to 1.5 μL and 8.1 μL, respectively, and the number of cycles was increased to 40 (Fig. 2). All PCR cocktails included a PCR negative control with nuclease-free water used instead of template DNA. Only when PCR negative controls showed no evidence of a target band did we proceed with the next steps (below). The PCR products of all eDNA samples showed clear target bands of appropriate size (133–140 bp). PCR of all field sampling negative controls showed no evidence of amplification. Because we observed non-target bands from PCR of some eDNA samples, we ran 20 μL of every PCR product from all eDNA samples in a 2% agarose gel and excised the target band manually with single-use scalpel blades. The gel containing the target fragment was purified with the Wizard SV Gel and PCR Clean-Up System (Promega, Madison, WI USA) following the manufacturer’s instructions.
Library preparation and sequencing
For eDNA samples where we successfully amplified the 16S rRNA fragment of target size, we employed a two-step tailed PCR approach to construct paired-end libraries and to assign unique identification of each sample for multiplexed sequencing (Fig. 2)34. The first-round PCR was performed to add primer-binding sequences for subsequent Illumina sequencing. In this step, the primer pairs contained both the degenerate primers above and the primer-binding sequences (Fig. 2; Supplementary Table S4). The 25 μL PCR cocktails contained 12.5 μL of 2 × Taq Mix (Froggabio), 9.25 μL of nuclease-free water, 0.875 μL of 10 μM each primer, 0.5 μL of 10 mg/ml BSA, and 1 μL of 0.1 ng/μL purified initial PCR product. The PCR cycling conditions were as follows: denaturation at 95 °C for 5 min followed by 18 cycles of 95 °C for 30 s, 68 °C for 30 s, 72 °C for 30 s and a final extension at 72 °C for 7 min. The PCR products were subsequently diluted 400 times and used in the second-round PCR as DNA template. The second-round PCR added Illumina adapter sequences and dual indices (combinations of D501-508 and D701-712) to identify different samples for sequencing on the HiSeq platform (Fig. 2; Supplementary Table S4). The 25 μL PCR cocktails contained 12.5 μL of 2 × Taq Mix (Froggabio), 9.26 μL of nuclease-free water, 0.87 μL of 10 μM each primer (Supplementary Table S4), 0.5 μL of 10 mg/mL BSA, and 1 μL of diluted first-round PCR product. The PCR cycling conditions were identical to the first round of PCR.
We used Solid Phase Reversible Immobilisation (SPRI) beads to separately purify 90 (15 sampling sites * 2 seasons * 3 PCR replicates) second-round PCR products, and then used the DeNovix dsDNA Ultra High Sensitivity Evaluation Kit (Wilmington, DE USA) to measure concentrations. Finally, 6 ng of DNA from each purified second-round PCR product were pooled and purified again using SPRI beads. The pooled samples were submitted to the Génome Québec Innovation Centre (Quebec, Canada) for 150 bp, paired-end run on the Illumina HiSeq 4000 platform.
Bioinformatics and data analysis
Quality control checks were performed on raw read data using FastQC v.0.11.8. No sequences were flagged as being of poor quality. We then used Trimmomatic v.0.3235 with “ILLUMINACLIP:HiSeq.adapter.fa:3:30:10:1 MINLEN:122” command under the paired-end mode to remove Illumina adapters36,37. After quality filtering and adapter removal, fastq files were converted to fasta files. We used VSEARCH v.2.5.038,39 to dereplicate identical reads (derep_fulllength), and then added the number of identical reads to the header line of the FASTA formatted data file (sizeout). Sequences with < 10 identical reads were removed (minuniquesize 10), reducing the potential for artifacts40.
Representative sequences with ≥ 10 identical reads were subjected to a nucleotide (NT) BLAST search (blastn, evalue 0.00001 and perc_identity 99)41,42 on NCBI using BLAST + v.2.7.143. The results were imported into MEGAN v.6.15.244, and taxa were assigned using the NCBI-NT Database and default lowest common ancestor (LCA) algorithm with Min Score = 200, Max Expected = 10−5, and Min Percent Identity = 99 to discard sequences that fell below our thresholds39,45.
We then manually tabulated the sequence abundance of each fish species based on the total number of identical reads for all sequences assigned to each fish species. Similar to Siegenthaler et al.46, we considered a species to have been detected if it was present in ≥ 2 PCR replicates. Species detected in only one PCR replicate sample were not considered further, making our results more conservative and less prone to reporting error39. We then generated rarefaction curves to assess sequencing coverage per sampling site and season, based on the mean normalized counts of taxonomic units detected with eDNA. A principal coordinates analysis (PCoA) was performed and Jaccard indices were calculated to visualize dissimilarity and similarity among sampling regions and seasons. The Ward’s Hierarchical clustering was used to identify which sites were more similar in species composition. The rarefaction curves, PCoA and clustering analysis were performed using the application ranacapa available at https://gauravsk.shinyapps.io/ranacapa. Bar graphs were generated in RStudio v.1.1.45647 with R v.3.5.348 using the package ggplot2 v.3.2.149 to visualize patterns of family and species diversity across sites and seasons. A heatmap was also created in RStudio using the diverse v.0.1.550 and foreign v.0.8–72 packages to visualize the community composition and spatial distribution using presence-absence data for taxonomic datasets.
Results
HiSeq sequencing results and initial analysis
The HiSeq paired-end sequencing yielded over 286 million paired-end reads. The read quality scores obtained were similar between libraries, with an average Phred score of 33. We employed stringent filtering parameters (as described above) and retained a high-confidence dataset consisting of ~ 10.1 million reads. The range in number of reads per library (water sample) was between 2168 and 1,413,298 (Supplementary Table S5). Overall, eDNA samples reached sufficient sequencing coverage based on the generated rarefaction curves (Supplementary Fig. S1).
Total fish species diversity
Initially a total of 101 fish species from 90 libraries was identified, and the taxonomic composition and read numbers were tabulated (Supplementary Table S5). Because these 90 libraries were obtained by using 3 PCR replicates for each of the 30 eDNA samples, and only species present in at least 2 PCR replicates were included in our analyses, we retained 75 fish species for all subsequent analyses, removing 26 fish species detected in only one PCR replicate (Supplementary Tables S6 and S5).
These 75 fish species belonged to 6 orders, 9 families, and 57 genera (Table 1). At the ordinal level, Cypriniformes had the highest diversity, comprising 56 species (75%), followed by 8 species of Siluriformes (11%), 4 species of each of Salmoniformes (5%) and Perciformes (5%), 2 species of Clupeiformes (3%), and 1 species of Beloniformes (1%) (Fig. 3a). Overall, we detected 4 pelagic species, 53 benthopelagic species (71% of all fish species detected), 17 demersal species, and one Cyprinidae hybrid species of unknown ecology; Dawkinsia tambraparniei (benthopelagic) had the lowest relative sequence abundance (21 reads), and was only identified in spring samples from Poyang Lake; Tachysurus nitidus (demersal) had the highest relative sequence abundance (3,639,140 reads) located in spring and summer samples in both the Tian-e-Zhou Reserve and Poyang Lake (Table 1). Our analysis suggested the presence of a number of non-native fish species including Atlantic salmon (Salmo salar), Bull trout (Salvelinus confluentus), Largemouth bass (Micropterus salmoides), Flathead chub (Platygobio gracilis) and Taiwanese salmon (Oncorhynchus formosanus), among others.

Total proportion of all fish species for each fish order identified (a) across seasons and regions, (b) within Tian-e-Zhou Reserve, and (c) within Poyang Lake.
Fish species diversity in different regions
In total, we identified 50 fish species in the Tian-e-Zhou Reserve, and 69 fish species in Poyang Lake (Tables 1 and 2). Of these, 44 species were common across the Tian-e-Zhou Reserve and Poyang Lake samples, which thus shared 59% of the total 75 fish species; 6 species were detected in the Tian-e-Zhou Reserve only, and 25 species were unique to Poyang Lake. We found significantly more fish species in Poyang Lake than in Tian-e-Zhou Reserve samples (2-way ANOVA with season and region as main effects: F(region) = 25.53, p < < 0.001).
The number of taxa clearly varied between the two regions. The fish species identified in the Tian-e-Zhou Reserve belonged to 5 orders, with 38 species (76%) of Cypriniformes followed by 4 species each of Salmoniformes (8%) and Siluriformes (8%), and 2 species each of Clupeiformes (4%) and Perciformes (4%) (Fig. 3b); the fish species identified in Poyang Lake belonged to 6 orders, with 51 species (74%) of Cypriniformes, followed by 8 species of Siluriformes (12%), 4 species of Perciformes (6%), 3 species of Salmoniformes (4%), 2 species of Clupeiformes (3%), and 1 species of Beloniformes (1%) (Fig. 3c). Beloniformes is a new order for the Tian-e-Zhou Reserve. Clearly in terms of species richness Cypriniformes dominated in both regions.
Fish species diversity in different seasons
In the Tian-e-Zhou Reserve, we detected 48 fish species in the spring and 34 fish species in the summer; in Poyang Lake, we detected 62 fish species in the spring and 58 fish species in the summer (Tables 1 and 2). Of these, 30 species were common to spring and summer samples from Tian-e-Zhou Reserve and Poyang Lake; 4 and 11 species were identified only in spring samples from Tian-e-Zhou Reserve and Poyang Lake, respectively, while 6 were identified only in summer samples from Poyang Lake; there were no fish species unique to the summer samples of the Tian-e-Zhou Reserve. For both regions, the detected number of species in the spring was greater than that of summer (2-way ANOVA with season and region as main effects: F(season) = 7.15, p = 0.013).
Jaccard similarity analysis indicated that species richness between spring and summer samples in Poyang Lake was most similar (0.74), species richness between spring and summer samples in Tian-e-Zhou Reserve was the third most similar (0.64), while species richness between summer samples of Tian-e-Zhou Reserve and spring samples of Poyang Lake were least similar (0.50; see Table 3). Overall, despite these seasonal differences, fish species composition of samples taken from the same water body were more similar than between.
Plots from our principal-coordinates analysis (PCoA) showed that spring samples from both regions, Tian-e-Zhou Reserve and Poyang Lake, were well separated from all other samples along the first PCoA axis explaining 31.3% of the variation in the data; summer samples were more similar and separated partially along the second PCoA axis explaining 18.6% of the variation in the data set (Fig. 4). This result was concordant with the result of our cluster analysis (Fig. 5).

Ordination analysis representing similarity in community composition based on eDNA taxonomic identification using Jaccard index.

Heatmap and cluster analysis of fish species composition of different samples in the Tian-e-Zhou Reserve and Poyang Lake in the spring and summer. Rows correspond to species IDs (see Table 1 or Supplementary Table S6), columns correspond to each sampling site and season. Bar colors refer to number of reads (normalized), with lighter colors indicating higher values (white represents absence). Cluster plot uses Ward's Hierarchical clustering method; sites with similar taxonomic composition cluster together. Color codes follow Fig. 4 (PCoA).
In the Tian-e-Zhou Reserve, Micropterus salmoides and Pseudolaubuca engraulis had the lowest and highest relative sequence abundance in spring samples respectively, changing in the summer samples to Spinibarbus denticulatus and Rhinogobius giurinus; in Poyang Lake, Dawkinsia tambraparniei and Tachysurus nitidus had the lowest and highest relative sequence abundance in spring samples, respectively, while again these switched to Pseudaspius leptocephalus and Rhinogobius giurinus in the summer samples (Table 1; Fig. 5). Overall, Pseudolaubuca engraulis, Rhinogobius giurinus, and Tachysurus nitidus had the highest relative sequence abundances.
Fish species diversity at different sampling sites
We identified different fish species richness in samples from different seasons at different sampling sites within regions. In general, the number of fish species detected in the spring sample was greater than that in the summer sample at each sampling site, except for sampling sites A, C, h, and j (Table 2). Overall, the number of species identified in the sampling sites from Poyang Lake in the spring and summer were higher than that in the sampling sites from Tian-e-Zhou Reserve in spring and summer, respectively. There were a few exceptions. For sampling site E from the Tian-e-Zhou Reserve, the spring sample contained higher richness than spring samples of most sampling sites in Poyang Lake. For sampling sites a and g from Poyang Lake, summer samples had lower richness than that of the summer samples of some sampling sites in Tian-e-Zhou Reserve (Table 2).
Across all 30 eDNA samples (with the exception of Tian-e-Zhou Reserve spring samples at sites A-D), we detected species inhabiting different water depths (pelagic, benthopelagic and demersal; Supplementary Fig. S2). For each sample, the number of benthopelagic fish species was highest, followed by demersal fishes and pelagic species. For the Tian-e-Zhou Reserve, 11 fish species were common to all sampling sites in the spring, and 10 species were common to all sampling sites in summer; in contrast, Poyang Lake had 17 and 6 species common to all sites in spring and summer, respectively (Supplementary Table S7). In total, 4 fish species were found in every sample in our study: Acrossocheilus kreyenbergii, Pseudolaubuca engraulis, Rhinogobius giurinus, and Tachysurus nitidus.
The relative abundance of fish species as reflected in sequence reads in the summer samples of the Tian-e-Zhou Reserve was similar to that of the summer samples in Poyang Lake, where most samples contained a similar dominant species, Rhinogobius giurinus, in high relative abundance (Supplementary Fig. S3). The dominant species in most spring samples of Tian-e-Zhou Reserve was Pseudolaubuca engraulis and was Tachysurus nitidus in most spring samples of Poyang Lake. In the Tian-e-Zhou Reserve spring samples, we recorded the highest number of reads for Cyprinidae, whereas Gobiidae had the highest number of reads in summer; in the spring samples of Poyang Lake, Bagridae appeared most abundant, switching to Cyprinidae and Gobiidae in the summer (Supplementary Fig. S4).
Prey fish of Yangtze finless porpoise
Of the 75 fish species detected using eDNA metabarcoding, 9 were known YFP prey fish species (Table 1). Three of the 9 prey species detected were common across all 30 eDNA samples among regions and seasons: Pseudolaubuca engraulis, Rhinogobius giurinus, and Tachysurus nitidus. The two regions generally shared the same YFP prey species, with Tian-e-Zhou Reserve containing one prey species not detected in Poyang Lake, and Poyang Lake similarly containing one prey species not found in the Tian-e-Zhou Reserve.
Discussion
Environmental DNA metabarcoding is increasingly employed to survey aquatic biodiversity, used successfully in lentic51, lotic15, estuarine52, coastal42 and deep-water marine systems53. Using this metabarcoding approach, we set out to design DNA primers that would allow us to assess the spatiotemporal dynamics of fish populations and communities across the Yangtze River Basin, including the 28 known prey fish species of the YFP. As would be expected given the geographic extent of our sampling and the taxonomic breadth of the prey species that we used to design our primers, we conservatively found the eDNA signatures of 75 fish species in the two regions that we surveyed across two seasons. Our eDNA investigations revealed differences in species richness between regions and seasons, and suggested that this method would be useful in mapping fish diversity in the Yangtze River and associated water bodies, including both introduced species (Table 1) and prey species of the YFP. Although we sampled from the water column typically one meter from the bottom for both water bodies (Supplementary Table S1), the fish identified with our eDNA metabarcoding approach were not just demersal but included benthopelagic and pelagic species as well.
Species diversity identified in different regions
Based on the presence of eDNA, we detected 19 more species in Poyang Lake than that in the Tian-e-Zhou Reserve overall. This is perhaps not surprising as Poyang Lake is markedly larger than the Tian-e-Zhou Reserve and is an open system, with inflows from the Ganjiang, Fuhe, Xinjiang, Raohe and Xiuhe Rivers, and connection to the Yangtze River via a channel54. In contrast, the Tian-e-Zhou Reserve is effectively isolated from the Yangtze River, only hydrologically linked through sluice gates during episodic flooding. Of the two focal regions in this study, previously published data on fish species that have been captured via traditional net survey methods allow for some comparisons with our eDNA survey data. In the Tian-e-Zhou Reserve, our eDNA surveys detected 10 fish species that had also been caught using fishing nets (Table 1): Gong et al.55 conducted net surveys and analysis of the year-round fish resources of the Tian-e-Zhou Reserve, capturing Carassius auratus, Chanodichthys erythropterus, Elopichthys bambusa, Hemiculter leucisculus, Hypophthalmichthys molitrix, Rhinogobius giurinus, Squalidus argentatus and Xenocypris davidi; additional net surveys by Gong et al.56 resulted in capture of another fish species, Pseudolaubuca engraulis; Acanthorhodeus chankaensis was further detected using net surveys in the Tian-e-zhou Reserve57. In Poyang Lake, we detected 31 fish species using eDNA that had also been caught using fishing nets (Table 1): Yang et al.58 and Jin et al.59 conducted net surveys of fish during spring, summer and autumn seasons, capturing 16 fish species; an additional, four fish species have been reported from Poyang Lake60,61,62 and 11 species in the broader Poyang Lake Basin54,63. Clearly future research should focus on the simultaneous comparisons of eDNA and net surveying within the same regions to directly assess sampling labor effort and accuracy; however, the fact that we detected many species reported from traditional net surveys gives us confidence that the patterns that we report here are real.
Fish species diversity in different seasons and at different sampling sites
We detected greater species richness in spring versus summer samples in both regions and at most of the sampling sites (Table 2). May through July encompasses the breeding season of most fish species in the Yangtze River64 and it is thus possible that detection probabilities were higher during our spring sampling simply because there were more eDNA sources for some species (e.g. gametes, fish fry)23. Although the species composition among sites in the same region had higher similarity (Table 3), changes in eDNA relative sequence abundance and detection of fish species at different sampling sites (Supplementary Fig. S3) may also reflect shifts in habitat use and distribution10,65. For example, Stoeckle et al.52 found that variation in fish eDNA detections reflected seasonal presence and habitat preferences determined using traditional surveys in coastal and estuarine marine habitats.
Prey fish of Yangtze finless porpoises
We found that three YFP prey fish species had among the highest number of total reads of any detected species (Pseudolaubuca engraulis—943,566, Rhinogobius giurinus—1,721,934 and Tachysurus nitidus—3,639,140), implying our metabarcoding approach may provide important insights into prey distributions and potentially relative abundances. Other researchers have found eDNA concentration to be positively related to field-measured density, biomass, or proportion of surveyed transects occupied in amphibians66, and fish67,68,69. Evans et al.13 further illustrated the potential of eDNA metabarcoding approaches for improving quantification of aquatic species diversity in natural environments and showed how eDNA metabarcoding can serve as an index of macrofaunal species abundance. With further comparisons between metabarcoding sequence read numbers and YFP prey species abundance, future conservation efforts could become more focused on managing or supplementing fish population numbers with hopefully positive consequences for YFP population persistence.
Sequence cut-offs and other caveats of our methodology
To our knowledge, sequence processing thresholds are not standardized for species diversity assessment using eDNA metabarcoding. For example, Miya et al.34 used 97% similarity to identify fish species, Valentini et al.51 used 98% similarity to identify amphibians and bony fish species, while Sato et al.41, Yamamoto et al.42, and Simmons et al.70 used 99% similarity to identify fish species. We explored the implications of sequence similarity cut-offs, comparing 97%, 98% and 99% similarity as our thresholds. Unsurprisingly, the results did vary depending on this cut-off with the number of fish operational taxonomic units (OTUs) detected increasing as we increased the threshold from 97 to 99%. Similarly, different authors have used different BLAST E-value (e.g. 10−5, 10−10 or 10−20)16,34,36. When we align a sequence to the nucleotide database for species identification, a higher sequence similarity, smaller BLAST E-value, and higher bit-score usually indicate sequence or species matches of higher quality44. We ultimately decided to use a 99% similarity cut-off, an E-value of 10−5, and set the lowest bit-score to 200. If there were to be internationally recognized values for these parameters for each gene and taxonomic group, the normalization of sequence processing steps could allow for more direct comparison of results between studies.
Our environmental DNA results imply the presence of fish species that have not yet been verified using traditional fishing methods, including a number of non-native species (Table 1). This is not surprising given the size of these water bodies, the sensitivity of eDNA surveys71,72, and the fact that aquaculture73 and the exotic fish trade74 are common in China. Two other considerations bear on our results. First, we used a single, short amplicon (133–140 bp) of the mitochondrial 16S rRNA gene where differences among closely related species may be insufficient to distinguish them. Second, definitive searches of on-line DNA data repositories like the NCBI-NT Database assume that data for all species are present and it is unlikely that there yet exists a complete catalogue of all native and non-native fish species for the Yangtze River Basin.
While we found differences between seasons in the number of species identified that may reflect life histories of local fish species and the use of different microhabitats over the annual cycle, these may also result from changes in the physicochemical properties of sampled water bodies. For example, elevated summer water temperatures can accelerate the rate of degradation of eDNA75. The trophic state of the water body also affects eDNA degradation; for example, eDNA decay rate of Cyprinus carpio was shown to be slowest in dystrophic water and fastest in oligotrophic water, and negatively correlated with dissolved organic carbon concentration76. Although our water samples showed little difference in temperature or dissolved oxygen between the Tian-e-Zhou Reserve or Poyang Lake (Supplementary Table S1), these abiotic factors did differ between seasons, possibly influencing not only fish diversity and behavior, but also eDNA production or persistence23. Further studies examining how aquatic abiotic parameters influence eDNA metabarcoding species detection and abundance estimations would be fruitful.
Another consideration for eDNA metabarcoding methods is the handling of negative controls. For example, we followed the protocol of Zou et al.77 and Fernández et al.78, only sequencing negative control samples if they showed evidence of positive PCR amplification. Our results were all negative, suggesting no contamination. However, other researchers30,39,42 regardless of visual confirmation of amplification, perform library preparations of negative controls for Illumina sequencing, using these data as a baseline to correct diversity metrics. Although it is often the case that negative control samples that show no PCR amplification also produce no (significant) Illumina sequencing reads79,80, researchers using these primers for eDNA metabarcoding fish species in the Yangtze River Basin should consider this more rigorous approach (sequencing negative control samples) when assessing amplification contamination.
Conclusion
While our intent was to develop metabarcoding tools to non-invasively survey fish biodiversity throughout the Yangtze River Basin, our approach also shows great promise for surveying prey fish species of the YFP. Our metabarcoding approach detected many fish species that have been documented using traditional sampling practices, implying greater fish species richness than traditional netting has suggested. The Chinese government once banned fishing during the spring fish spawning season in certain areas of the Yangtze River only81 but now has implemented a complete 10-year fishing ban in key areas of the Yangtze River because of dramatic declines in diversity and abundance of aquatic taxa82. The Yangtze River is the largest river in East Asia, and the human population along its watershed exceeds 450 million people83. Environmental DNA metabarcoding could help fill an important but missing knowledge gap for freshwater biodiversity in China, and assist in future conservation planning across much of the country.
Data availability
The authors confirm all data will be deposited into FigShare upon manuscript acceptance.
References
Gangloff, M. M., Edgar, G. J. & Wilson, B. Imperilled species in aquatic ecosystems: emerging threats, management and future prognosis. Aquatic Conserv. Mar. Freshw. Ecosyst. 26, 858–871 (2016).
WWF. Living Planet Report—2018: Aiming Higher (eds Grooten, M. & Almond, R.E.A.). WWF, Gland, Switzerland (2018).
Zhou, X. et al. Population genomics of finless porpoises reveal an incipient cetacean species adapted to freshwater. Nat. Commun. 9, 1276 (2018).
Mei, Z. et al. Accelerating population decline of Yangtze finless porpoise (Neophocaena asiaeorientalis asiaeorientalis). Biol. Conserv. 153, 192–200 (2012).
Wang, D., Turvey, S.T., Zhao, X. & Mei, Z. Neophocaena asiaeorientalisssp.asiaeorientalis. The IUCN Red List of Threatened Species 2013 e.T43205774A45893487, http://dx.doi.org/https://doi.org/10.2305/IUCN.UK.2013-1.RLTS.T43205774A45893487.en(2013).
Yang, J. et al. A preliminary study on diet of the Yangtze finless porpoise using next-generation sequencing techniques. Mar. Mammal Sci. 35, 1579–1586 (2019).
Wang, D. Population status, threats and conservation of the Yangtze finless porpoise. Chin. Sci. Bull. 54, 3473–3484 (2009).
Wu, J. et al. Progress in studies on water ecology in Tian’e Zhou Oxbow. Acta Hydrobiol. Sin. 41, 935–946 (2017) (In Chinese).
Nabi, G., Hao, Y., Robeck, T. R., Zheng, J. & Wang, D. Physiological consequences of biologic state and habitat dynamics on the critically endangered Yangtze finless porpoises (Neophocaena asiaeorientalis ssp. asiaeorientalis) dwelling in the wild and semi-natural environment. Conserv. Physiol. 6, coy072 (2018).
Stewart, K., Ma, H., Zheng, J. & Zhao, J. Using environmental DNA to assess population-wide spatiotemporal reserve use. Conserv. Biol. 31, 1173–1182 (2017).
Chen, M. et al. Parentage-based group composition and dispersal pattern studies of the Yangtze Finless Porpoise population in Poyang Lake. Int. J. Mol. Sci. 17, 1268 (2016).
Civade, R. et al. Spatial representativeness of environmental DNA metabarcoding signal for fish biodiversity assessment in a natural freshwater system. PLoS ONE 11, e0157366 (2016).
Evans, N. T. et al. Quantification of mesocosm fish and amphibian species diversity via environmental DNA metabarcoding. Mol. Ecol. Resour. 16, 29–41 (2016).
Hänfling, B. et al. Environmental DNA metabarcoding of lake fish communities reflects long-term data from established survey methods. Mol. Ecol. 25, 3101–3119 (2016).
Fujii, K. et al. Environmental DNA metabarcoding for fish community analysis in backwater lakes: a comparison of capture methods. PLoS ONE 14, e0210357 (2019).
Andruszkiewicz, E. A. et al. Biomonitoring of marine vertebrates in Monterey Bay using eDNA metabarcoding. PLoS ONE 12, e0176343 (2017).
Harper, L. R. et al. Needle in a haystack? A comparison of eDNA metabarcoding and targeted qPCR for detection of the great crested newt (Triturus cristatus). Ecol. Evol. 8, 6330–6341 (2018).
Günther, B., Knebelsberger, T., Neumann, H., Laakmann, S. & Arbizu, P. M. Metabarcoding of marine environmental DNA based on mitochondrial and nuclear genes. Sci. Rep. 8, 14822 (2018).
Djurhuus, A. et al. Evaluation of marine zooplankton community structure through environmental DNA metabarcoding. Limnol. Oceanogr. Methods 16, 209–221 (2018).
Ficetola, G. F., Miaud, C., Pompanon, F. & Taberlet, P. Species detection using environmental DNA from water samples. Biol. Lett. 4, 423–425 (2008).
Taberlet, P., Coissac, E., Hajibabaei, M. & Rieseberg, L. H. Environmental DNA. Mol. Ecol. 21, 1789–1793 (2012).
Thomsen, P. F. et al. Monitoring endangered freshwater biodiversity using environmental DNA. Mol. Ecol. 21, 2565–2573 (2012).
Stewart, K. A. Understanding the biotic and abiotic factors on sources of aquatic environmental DNA. Biodivers. Conserv. 28, 983–1001 (2019).
Lopes, C. M. et al. eDNA metabarcoding: a promising method for anuran surveys in highly diverse tropical forests. Mol. Ecol. Resour. 17, 904–914 (2017).
Hobbs, J., Helbing, C. C. & Veldhoen, N. Environmental DNA protocol for freshwater aquatic ecosystems version 2.2. Report for the BC Ministry of Environment, Victoria BC Canada (2017).
Laramie, M.B., Pilliod, D.S., Goldberg, C.S. & Strickler, K.M. Environmental DNA sampling protocol—Filtering water to capture DNA from aquatic organisms.U.S. Geological Survey Techniques and Methods, book 2, chap. A13, 15 p. (2015).
Ma, H. et al. Characterization, optimization, and validation of environmental DNA (eDNA) markers to detect an endangered aquatic mammal. Conserv. Genet. Resour. 8, 561–568 (2016).
Thomsen, P. F. et al. Detection of a diverse marine fish fauna using environmental DNA from seawater samples. PLoS ONE 7, e41732 (2012).
Dougherty, M. M. et al. Environmental DNA (eDNA) detects the invasive rusty crayfish Orconectes rusticus at low abundances. J. Appl. Ecol. 53, 722–732 (2016).
Evans, N. T. et al. Fish community assessment with eDNA metabarcoding: effects of sampling design and bioinformatic filtering. Can. J. Fish. Aquat. Sci. 74, 1362–1374 (2017).
Renshaw, M. A., Olds, B. P., Jerde, C. L., Mcveigh, M. M. & Lodge, D. M. The room temperature preservation of filtered environmental DNA samples and assimilation into a phenol-chloroform-isoamyl alcohol DNA extraction. Mol. Ecol. Resour. 15, 168–176 (2015).
Thompson, J. D., Higgins, D. G. & Gibson, T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680 (1994).
Hall, T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Sym. Ser. 41, 95–98 (1999).
Miya, M. et al. MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: detection of more than 230 subtropical marine species. R. Soc. Open Sci. 2, 150088 (2015).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Olds, B. P. et al. Estimating species richness using environmental DNA. Ecol. Evol. 6, 4214–4226 (2016).
Deiner, K. et al. Long-range PCR allows sequencing of mitochondrial genomes from environmental DNA. Methods Ecol. Evol. 8, 1888–1898 (2017).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584 (2016).
Port, J. A. et al. Assessing vertebrate biodiversity in a kelp forest ecosystem using environmental DNA. Mol. Ecol. 25, 527–541 (2016).
Pochon, X., Zaiko, A., Fletcher, L. M., Laroche, O. & Wood, S. A. Wanted dead or alive? Using metabarcoding of environmental DNA and RNA to distinguish living assemblages for biosecurity applications. PLoS ONE 12, e0187636 (2017).
Sato, H., Sogo, Y., Doi, H. & Yamanaka, H. Usefulness and limitations of sample pooling for environmental DNA metabarcoding of freshwater fish communities. Sci. Rep. 7, 14860 (2017).
Yamamoto, S. et al. Environmental DNA metabarcoding reveals local fish communities in a species-rich coastal sea. Sci. Rep. 7, 40368 (2017).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinform. 10, 421 (2009).
Huson, D. H. et al. MEGAN Community Edition—interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 12, e1004957 (2016).
DiBattista, J. D. et al. Assessing the utility of eDNA as a tool to survey reef-fish communities in the Red Sea. Coral Reefs 36, 1245–1252 (2017).
Siegenthaler, A. et al. Metabarcoding of shrimp stomach content: Harnessing a natural sampler for fish biodiversity monitoring. Mol. Ecol. Resour. 19, 206–220 (2019).
Racine, J. S. RStudio: a platform-independent IDE for R and Sweave. J. Appl. Econ. 27, 167–172 (2012).
R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org (2018).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis (eds Gentleman, R., Hornik, K. & Parmigiani, G.) 1–212 (Springer, 2009).
Guevara, M. R., Hartmann, D. & Mendoza, M. diverse: an R package to analyze diversity in complex systems. R J. 8, 60–78 (2016).
Valentini, A. et al. Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Mol. Ecol. 25, 929–942 (2016).
Stoeckle, M. Y., Soboleva, L. & Charlop-Powers, Z. Aquatic environmental DNA detects seasonal fish abundance and habitat preference in an urban estuary. PLoS ONE 12, e0175186 (2017).
Thomsen, P. F. et al. Environmental DNA from seawater samples correlate with trawl catches of subarctic, deepwater fishes. PLoS ONE 11, e0165252 (2016).
Huang, L., Wu, Z. & Li, J. Fish fauna, biogeography and conservation of freshwater fish in Poyang Lake Basin China. Environ. Biol. Fish. 96, 1229–1243 (2013).
Gong, J. et al. Interannual variation of the fish community structure in the Tian-e-Zhou Oxbow of Yangtze River. J. Hydroecol. 39, 46–53 (2018) (In Chinese).
Gong, C., Chen, Z. & Cheng, F. The status and management suggestions of the Yangtze finless porpoise prey fish in the Tian-e-Zhou Oxbow of Yangtze River. China Fish. 6, 43–45 (2019) (In Chinese).
Wang, T., Wang, H., Sun, G., Huang, D. & Shen, J. Length–weight and length–length relationships for some Yangtze River fishes in Tian-e-zhou Oxbow China. J. Appl. Ichthyol. 28, 660–662 (2012).
Yang, S., Li, M., Zhu, Q., Wang, M. & Liu, H. Spatial and temporal variations of fish assemblages in Poyanghu Lake. Resour. Environ. Yangtze Basin 24, 54–64 (2015) (In Chinese).
Jin, B. et al. Fish assemblage structure in relation to seasonal environmental variation in sub-lakes of the Poyang Lake floodplain, China. Fish. Manag. Ecol. 26, 131–140 (2019).
Fang, C. et al. Fish resources in Poyang Lake and their utilization. Jiangsu Agric. Sci. 44, 233–243 (2016) (In Chinese).
Zhong, B. et al. Classification of Pelteobagrus fish in Poyang Lake based on mitochondrial COI gene sequence. Mitochondrial DNA A 27, 4635–4637 (2016).
Xiong, G., Zhang, T., Lin, Y., Wang, W. & You, X. Analysis of some characters of fish in the inner Lake of Poyang Lake Wetland. Jiangxi Fish. Sci. Technol. 3, 10–12 (2018) (In Chinese).
Liu, X. et al. Biodiversity pattern of fish assemblages in Poyang Lake Basin: threat and conservation. Ecol. Evol. 9, 11672–11683 (2019).
Liu, M. et al. Species diversity of drifting fish eggs in the Yangtze River using molecular identification. PeerJ 6, e5807 (2018).
Hinlo, R., Furlan, E., Suitor, L. & Gleeson, D. Environmental DNA monitoring and management of invasive fish: comparison of eDNA and fyke netting. Manag. Biol. Invasion 8, 89–100 (2017).
Pilliod, D. S., Goldberg, C. S., Arkle, R. S. & Waits, L. P. Estimating occupancy and abundance of stream amphibians using environmental DNA from filtered water samples. Can. J. Fish. Aquat. Sci. 70, 1123–1130 (2013).
Lacoursière-Roussel, A., Côté, G., Leclerc, V. & Bernatchez, L. Quantifying relative fish abundance with eDNA: a promising tool for fisheries management. J. Appl. Ecol. 53, 1148–1157 (2016).
Lacoursière-Roussel, A., Rosabal, M. & Bernatchez, L. Estimating fish abundance and biomass from eDNA concentrations: variability among capture methods and environmental conditions. Mol. Ecol. Resour. 16, 1401–1414 (2016).
Ushio, M. et al. Quantitative monitoring of multispecies fish environmental DNA using high-throughput sequencing. Metabarcod. Metagenom. 2, 1–15 (2018).
Simmons, M., Tucker, A., Chadderton, W. L., Jerde, C. L. & Mahon, A. R. Active and passive environmental DNA surveillance of aquatic invasive species. Can. J. Fish. Aquat. Sci. 73, 76–83 (2016).
Smart, A. S., Tingley, R., Weeks, A. R., vanRooyen, A. R. & McCarthy, M. A. Environmental DNA sampling is more sensitive than a traditional survey technique for detecting an aquatic invader. Ecol. Appl. 25, 1944–1952 (2015).
Eiler, A., Löfgren, A., Hjerne, O., Nordén, S. & Saetre, P. Environmental DNA (eDNA) detects the pool frog (Pelophylax lessonae) at times when traditional monitoring methods are insensitive. Sci. Rep. 8, 5452 (2018).
Lin, Y., Gao, Z. & Zhao, A. Introduction and use of non-native species for aquaculture in China: status, risks and management solutions. Rev. Aquacult. 7, 28–38 (2015).
Xiong, et al. Non-native freshwater fish species in China. Rev. Fish. Biol. Fish. 25, 651–687 (2015).
Pilliod, D. S., Goldberg, C. S., Arkle, R. S. & Waits, L. P. Factors influencing detection of eDNA from a stream-dwelling amphibian. Mol. Ecol. Resour. 14, 109–116 (2014).
Eichmiller, J. J., Best, S. E. & Sorensen, P. W. Effects of temperature and trophic state on degradation of environmental DNA in lake water. Environ. Sci. Technol. 50, 1859–1867 (2016).
Zou, K. et al. eDNA metabarcoding as a promising conservation tool for monitoring fish diversity in a coastal wetland of the Pearl River Estuary compared to bottom trawling. Sci. Total Environ. 702, 134704 (2020).
Fernández, S., Rodríguez-Martínez, S., Martínez, J. L., Garcia-Vazquez, E. & Ardura, A. How can eDNA contribute in riverine macroinvertebrate assessment? A metabarcoding approach in the Nalón River (Asturias, Northern Spain). Environ. DNA 1, 385–401 (2019).
Lacoursière-Roussel, A. et al. eDNA metabarcoding as a new surveillance approach for coastal Arctic biodiversity. Ecol. Evol. 8, 7763–7777 (2018).
Liu, Y. et al. Application of environmental DNA metabarcoding to spatiotemporal finfish community assessment in a temperate embayment. Front. Mar. Sci. 6, 674 (2019).
Xie, X. et al. Are river protected areas sufficient for fish conservation? Implications from large-scale hydroacoustic surveys in the middle reach of the Yangtze River. BMC Ecol. 19, 42 (2019).
Xinhua. China starts 10-year fishing ban on Yangtze River.China Daily;https://www.chinadaily.com.cn/a/202001/02/WS5e0d4851a310cf3e35581f65.html(2020).
Yang, S., Xu, K., Milliman, J. D. & Wu, C. Decline of Yangtze River water and sediment discharge: Impact from natural and anthropogenic changes. Sci. Rep. 5, 12581 (2015).
Acknowledgements
The authors thank the staff of the Tian-e-Zhou Reserve for providing help in sampling, and Wenxi Feng and Aaron Sneep from Queen’s University for assistance in making the map. The authors would also like to thank the National Natural Sciences Foundation of China (KAS, 31400468), the National Key Programme of Research and Development of Ministry of Science and Technology of China (JZ, 2016YFC0503200), the National Key Experimental Instrument Research and Development Project of China (LM, 0400231024), the Tongji University State Key Laboratory for Pollution Control and Resource Reuse (KAS), the Chinese Scholarship Council (CQ), and the Natural Sciences and Engineering Research Council of Canada (Discovery Grant to SCL) for financially supporting this research.
Author information
Authors and Affiliations
Contributions
C.Q. collected the water samples and performed the laboratory work. K.A.S. designed the sampling regime. R.C.C. and S.C.L. contributed in the laboratory work, and Q.C., K.A.S., R.C.C., and S.C.L. contributed to data analysis, and manuscript writing and editing. J.Z. assisted in the collection of samples. C.G. assisted in the collection of samples in the Tian-e-Zhou Reserve. R.C.C., S.C.L., Y.W., L.M., and J.Z. assisted in the study design.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qu, C., Stewart, K.A., Clemente-Carvalho, R. et al. Comparing fish prey diversity for a critically endangered aquatic mammal in a reserve and the wild using eDNA metabarcoding. Sci Rep 10, 16715 (2020). https://doi.org/10.1038/s41598-020-73648-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-73648-2
This article is cited by
-
Application of eDNA metabarcoding for monitoring the fish diversity of the Jiangjin to Fuling section of the upper reaches of the Yangtze River
Hydrobiologia (2023)
-
eDNA metabarcoding as a promising conservation tool to monitor fish diversity in Beijing water systems compared with ground cages
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
