
Abstract
Breeding studies in walnut (Juglans regia L.) are usually time consuming due to the long juvenile period and therefore, this study aimed to determine markers associated with time of leaf budburst and flowering-related traits by performing a genome-wide association study (GWAS). We investigated genotypic variation and its association with time of leaf budburst and flowering-related traits in 188 walnut accessions. Phenotypic data was obtained from 13 different traits during 3 consecutive years. We used DArT-seq for genotyping with a total of 33,519 (14,761 SNP and 18,758 DArT) markers for genome-wide associations to identify marker underlying these traits. Significant correlations were determined among the 13 different traits. Linkage disequilibrium decayed very quickly in walnut in comparison with other plants. Sixteen quantitative trait loci (QTL) with major effects (R2 between 0.08 and 0.23) were found to be associated with a minimum of two phenotypic traits each. Of these QTL, QTL05 had the maximum number of associated traits (seven). Our study is GWAS for time of leaf budburst and flowering-related traits in Juglans regia L. and has a strong potential to efficiently implement the identified QTL in walnut breeding programs.
Similar content being viewed by others


Introduction
The genus Juglans consists of more than 20 species and is widely distributed worldwide1. Juglans regia L. is known as common walnut (English or Persian walnut), and is the most commercially important species in the genus due to its edible and highly nutritious nuts. Walnut is long-lived, deciduous, monoecious, open-pollinated and generally dichogamous2. J. regia is a diploid species with 16 haploid chromosomes3 and has an estimated genome size of ~ 606 Mb4. Genome of J. regia was previously reported by several authors5,6,7,8,9. Turkey has suitable ecological conditions for walnut cultivation and it produced about 5.5% (210,000 tons) of the total world production of 3,829,626 tons in 201710.
Advanced molecular methods have revolutionized population genetics and evolutionary biology studies in different plants. The development of high-density DNA markers has played an important role in understanding the genetic diversity of different germplasm and in accelerating breeding programs and selection efficiency for complex traits in plants. Unfortunately, to date, there are few studies aiming to develop high throughput molecular marker systems to characterize walnut populations11,12,13,14,15,16,17. Walnut is a perennial species with a long juvenile period resulting in the time-consuming breeding cycle. Walnut breeding would therefore greatly benefit from the development of molecular markers that could be used for diversity assessment, gene discovery, marker-assisted selection, genomic selection, and other breeding applications. Over the past two decades, single nucleotide polymorphism (SNP) markers have become the ultimate choice to characterize germplasm due to the possibility of developing thousands of SNPs in a considerably short time using next-generation sequencing technologies (NGS).
Genotyping by sequencing (GBS) has been developed to make simultaneous SNP discovery and genotyping as a rapid and robust method for reduced-representation sequencing of multiplexed samples that combines genome-wide molecular marker discovery and genotyping18. The Diversity Arrays Technology (DArT) is a cost-effective sequence-independent ultra-high-throughput marker system. This technology has developed a GBS platform known as DArT-seq, which provides an opportunity to select genome fractions corresponding predominantly to active genes. Restriction enzymes used in this method separate low copy sequences that informative for marker discovery from the repetitive fraction of the genome. Then, representative fragments are sequenced on Next Generation Sequencing (NGS) platforms. As a result, DArTseq offers affordable genome profiling by producing high-density SNPs as well as markers of presence and absence19.
In plant breeding, one of the main aims is to determine the genetic dissection of quantitative traits. In this regard, quantitative trait loci (QTL) that are affecting the observed phenotype can be detected by using linkage mapping and linkage disequilibrium analysis. Linkage mapping is restricted to a relatively low genomic resolution when evaluating the recombination events within the mapping populations. An alternative is genome-wide association mapping (GWAS) that exploits linkage disequilibrium (LD) in diverse populations to find putative QTL. This method has several advantages over linkage mapping as it uses natural variation, and has higher genetic resolution due to exploiting historical recombinations20. Recently, GWAS was also applied to walnut for ecophysiological traits such as water use efficiency as estimated by carbon isotope discrimination, and photosynthetic capacity17,21 as well as phenology, yield and lateral bearing traits14,15; while several other studies reported a few QTLs associated with nut-related traits12,16. However, QTL studies in walnuts are limited when it compared to other plant species.
For perennial fruit species, in the context of global warming, endodormancy release may be a critical step in the future due to insufficient chill accumulation, directly affecting flowering quality and uniformity, and thus leading to a drastic reduction of fruit production22. Walnut tree, like the other decidious tree species, requires winter chilling and heat the breaking of dormancy in the spring23. On the other hand, time of budburst and leafing date traits are very important in walnut cultivation due to spring frost that may cause significant losses in the production in some years especially in Turkey where it is prevalent24. Flowering-related traits are key factors not only in the plant life cycle but also in determining the productivity for walnut. The female flower buds can bear at the terminal and lateral shoots of walnut depending on cultivar. Particularly, the lateral bud flowering, nut setting types and female flower abundance are one of the most important features in walnut breeding programs due to association with high yield24,25. In addition, syncronizing the flowering habit of males and females is another important issue in terms of fertilization biology in a walnut orchard. Furthermore, high heritability were indicated for leafing date, heterodichogamy and female/male blooming were in walnut14,26. Therefore, the development of molecular markers related to phenology and flowering traits in walnut will play an important role in future breeding programs and will facilitate the development of other morphological, nutritional and physiological characteristics without being confounded by flowering time differences among individuals.
In this study, we have investigated the walnut population showing significant variations in terms of leaf budburst and flowering-related traits located in the Kahramanmaraş province, which is one of the walnut growing areas in Turkey, to (1) understand the genetic diversity and structure of walnut population and study its linkage disequilibrium, and (2) identify associations between molecular markers with time of leaf budburst and flowering-related traits using genome-wide association mapping to aid marker-assisted breeding in walnut. Besides, this study is the first study to identify markers associated with several yield-related traits such as catkin abundance, female flower abundance, nut setting type, and inflorescence habit in walnut.
Materials and methods
Plant materials
The walnut collection and breeding studies carried out in the Nuts Application and Research Center (SEKAMER), Kahramanmaraş, Turkey. The SEKAMER is located at 37° 35′ 27ʺ N latitude, 37° 03′ 28ʺ E longitude and 930 m above the sea level. Kahramanmaraş has a mild climate between the Mediterranean and continental with 727 mm yearly precipitation and 16.9 °C average yearly temperature. In this study, a collection of 188 walnut accessions (Supplementary Table S1) in SEKAMER were used for association mapping. The 188 walnut accessions were selected from approximately 1,200 accessions in the germplasm based on their difference from each other in terms of their phenology, yield and nut related traits as well as their age. The accessions were at 9–12 ages during this study.
Phenotyping
A total of 13 phenological traits were observed during the budburst and flowering period in the 2016, 2017 and 2018 growing seasons for 188 walnut accessions. The accessions were characterized based on the Descriptor for Walnut27 and the International Union for the Protection of New Varieties of Plants28. A list of characteristics and their definitions were presented in Table 1. The observation of phenotypic data as dates was recorded as the number of Julian days from January 1st of each year.
DNA extraction and genotyping
Collected leaves were washed with distilled water in order to clean dust, frozen in liquid nitrogen, and stored at − 80 °C until DNA extractions. Genomic DNA extraction was carried out according to the procedure described29 with minor modifications30. The purity and quantity of extracted DNA were measured using agarose gel electrophoresis and Qubit fluorometer (Invitrogen) according to the manufacturer's protocol. DNA concentrations of all accessions were fixed to 80–100 ng/µl for DArT-seq analysis.
DNA samples were processed in digestion/ligation reactions for complexity reduction using PstI-MseI as described31 to generate the DArT-seq markers. DArTSeq markers with MAF < 5% and missing data > 20% were discarded from all analyses. The physical positions of the DArT-seq markers were obtained by aligning marker sequences on the walnut reference genome5. BLAST+ software (https://www.ncbi.nlm.nih.gov/bookshelf/br.fcgi?book=helpblast) was used to adentify the physical positions of the markers with word size equal to 20 and evalue < 1e − 10. For sequences with multiple hits on the genome, the hit with the smallest evalue was considered as the physical position.
Statistical analysis
Pairwise correlations between different phenotypes were calculated using R (https://www.r-project.org). The hierarchal clustering of phenotypes and individuals using the phenotypic data was calculated using Euclidean distance and was plotted using the R function ‘heatmap.2’. SNPs with known scaffold positions were used to estimate the LD between markers using the squared allele frequency correlation (R2) following Hill and Weir32. Pairwise R2 values among SNPs located on the same scaffold were plotted against the physical distance (bp) between both SNPs to estimate the LD decay. The second-degree loess smoothing was calculated and plotted using R. Population structure was defined using the software ADMIXTURE33. The analysis was run with k value ranging from 2 to 20 and ten cross validations33. To avoid bias due to the linkage disequilibrium, we used PLINK34 software to prune SNPs at r2 value of 0.5. We ran 100 replicates of the ADMIXTURE analysis for each k. The most probable k was defined as the smallest k value in which the cross validation values of the hundred replicates had no significant difference from that for the next k.
Genome-wide association study
Restricted maximum likelihood (REML) analysis was used to calculate the narrow sense heritability by fitting the genotypic and phenotypic data as described in Yang et al.35. The mixed linear model (MLM) implemented in the genome association and prediction integrated tool (GAPIT) R package was used for the GWAS analysis36. The same package calculates the principal component (PC) analysis and the genomic relatedness matrix using VanRaden37 method and fits them as covariates for the GWAS analysis. The Bayesian information criterion (BIC) was used to find the optimal number of PCs to be fitted in the GWAS analysis for each trait. Bonferroni38 test was used to declare significant threshold at p < 0.05 for the GWAS analysis. Markers detected at this threshold were declared as highly significant markers. However, because of the stringency of Bonferroni test that ignores linkage disequilibrium and assumes independent markers, we reported all DArT-seq markers with p < 10−4 as suggestive associations if they had high linkage disequilibrium with a highly significant marker associated with a multi-trait QTL (defined when a marker is associated with several different traits). The full scaffold ordering information was obtained from Kefayati39 in which they used an F1 population with 175 individuals from a cross between Chandler and Kaplan-86 walnut cultivars to develop a genetic map. Predicted genes were extracted from the genome if they had a distance smaller than 20 kb from the associated marker. This value was selected based on the linkage disequilibium results.
Results
Phenotypic variations and correlation between phenotypic traits
In the present study, the data for 13 phenological traits related to leaf budburst and flowering were collected during three growing seasons (2016, 2017 and 2018). Phenological data related to leaf budburst and flowering traits are presented in Supplementary Table S2. Significant variations were obtained in most of the studied phenological traits. The budbreak date ranged from 67 (2018) to 109 (2017) days, while leafing time ranged from 76 (2018) to 117 (2017) days. While the first and last female blooming dates were 104.96 and 117.51 Julian days, the first and last male blooming dates were 102.05 and 109.03 Julian days on average, respectively. Female flowering period of walnut accessions varied from 4 to 25 days, and male flowering period varied between 2 and 15 days during three years. Female abundance, fruit setting type, and lateral bud flowering are important yield parameters in walnut. Walnut accessions were approximately ‘moderate’ in terms of female flower abundance (5.65) and catkin abundance (4.78) on average. Identification of ‘solitary’ and ‘bunchy’ walnut genotypes in all vegetation periods was an important source of variation for fruit setting type. Each walnut accessions had different inflorescence habits, while ‘protandrous’ was predominant. The average percentage of lateral bud flowering was 64.7%. While seasonal shifts in phenological findings were observed between the years, consistent findings were obtained between walnut accessions during three consecutive years. In addition, the estimation of narrow-sense heritabilities ranged from 12.3 to 95.4% for the average of traits across the three growing seasons suggesting that the considerable proportion of the phenotypic variation for most of the evaluated traits can be explained by genetic factors. High narrow-sense heritabilities (h2) were obtained for the budburst (0.877) and leafing times (0.888) as well as for the first (0.936 and 0.715) and the last (0.846 and 0.611) female and male blooming dates, respectively. The h2 values for inflorescence habit (0.552), lateral bud flowering (0.538) and female abundance were intermediate (0.455), while the values for female (0.123) and male (0.136) flowering periods, catkin abundance (0.188) and nut setting type (0.147) were low.
Many traits related to flowering and time of leaf budburst showed significant correlations (Fig. 1; Supplementary Data 1). Highly significant positive correlations were observed between budburst and leafing times with other traits, except nut setting type and female and male flowering periods, but both traits were negatively correlated with male flowering period and catkin abundance. The first and the last female blooming dates had positive correlations with female flowering period (0.30 and 0.59), while they had significantly negative correlations with catkin abundance (− 0.32 and − 0.35) and inflorescence habit (− 0.43 and − 0.35) at p = 0.001. The first and the last male blooming dates were correlated positively with female abundance (0.33 and 0.33), inflorescence habit (0.68 and 0.68) and lateral bud flowering traits (0.35 and 0.37), while the first male blooming dates had negative correlations with catkin abundance (− 0.28) and male flowering period (− 0.32) at p = 0.001. Female flowering period correlated significantly and positively with female abundance (0.37) and lateral bud flowering traits (0.41) at p = 0.001. However, it was correlated negatively with catkin abundance at p = 0.001 (− 0.28) and male flowering period at p = 0.01 (− 0.20). There was a positive correlation between male flowering period and catkin abundance (0.52) at p = 0.001. Female abundance and lateral bud flowering traits had significant positive correlations with all studied traits except male flowering period and catkin abundance. The nut setting type was positively correlated with female flower abundance (0.29) and lateral bud flowering (0.30) at p = 0.001. In addition to these results, inflorescence habit was positively correlated with lateral bud flowering at p = 0.001 (0.26).

Correlation analysis results of phenological characteristics related to time of leaf budburst and flowering-related traits.
Phenotypic and genotypic diversity and linkage disequilibrium in the walnut germplasm
The 13 phenotypic traits were grouped into eight clusters. The first one includes flowering period of male flowers and catkin abundance, while the second one contains time of leaf budburst and leafing time. The first and the last blooming dates of female flowers were in the third cluster, while inflorescence habit was in the fourth cluster. The first and the last blooming dates of male flowers were in the fifth cluster, while nut seting type and flowering period of female flowers were in sixth and seventh clusters, respectively. The last cluster included female flower abundance and lateral bud flowering traits (Fig. 2a). Clustering the germplasm based on the 13 phenotypic traits divided it into three distinguished clusters (Fig. 2a). Cluster one (C1) generally had phenotypic values below (blue colour in Fig. 2a) the average for all traits except male flowering period (7.7 vs 6.8 for C2 and C3) and catkin abundance (5.3 vs 4.4 for C2 and C3) in which it showed above average phenotypic response (red colour in Fig. 2a). Cluster two and cluster three (C2 and C3) are more similar to each other than cluster one. For C2 and C3, above average phenotypic values were observed for the traits time of budburst (88.3 vs 84.3 for C1), leafing time (98 vs 94.2 for C1), female flowering period (12.9 vs 11.7 for C1), female abundance (6.1 vs 4.9 for C1) and lateral bud flowering (74 vs 49 for C1); and values were below average for male flowering period and catkin abundance, and variable responses for nut setting type. However, C2 had phenotypic values above the average for traits the first and the last male blooming dates and inflorescence habit and values below the average for the first and the last female blooming dates were in contrast to C3 (Fig. 2a).

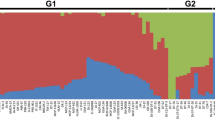
(a) Phenotypic clustering among 13 phenological characteristics reletad to flowering measured and population sutrucutere in 188 walnut genotypes; (b) population structure results for k = 2; (c) population structure results for k = 10; (d) clustering among genotypes based on the kinship relatedness matrix and their relation to the phenotype-based clustering.
In this study, a total of 23,029 DArT-seq derived SNPs and 30,031 DArT markers were identified, and were reduced to a total of 33,519 (14,761 SNP and 18,758 DArT) markers after filtering out markers with low allele frequency (< 5%) and high missing data (> 20%). Population structure analysis showed that 10 (Fig. 2c) could be a reasonable estimation of the number of ancestral subpopulations as the CV values for the 100 replicates of k = 10 was not significantly different from that for k = 11. PCA analysis also confirmed the presence of complex structure in our gemrplasm with multiple clusters distributed along the first three PCs, which explained 10.4%, 6.3% and 4.4% of the total variation, respectively (Supplementary Fig. S1). However, at k = 2 (Fig. 2b), the majority of the phenotypic C1 cluster formed one subpopulation with considerable admixture with the other subpopulation. Almost 82.6% of C1 was from Q1 (blue) and 17.4% from Q2 (green), while C2 and C3 were 36.2% from Q1 and 63.8% from Q2. Similarly, the dendrogram based on the kinship relatedness isolated large proportion of the phenotypic C1 in one group (Fig. 2d; Supplementary Fig. S2).
The walnut germplasm showed rapid LD decay with small LD blocks indicating very large ancestral effective population size (Supplementary Fig. S3). Using SNPs that exists on the same contig, LD decayed below r2 value of 0.2 at 16.6 kb and below r2 value of 0.15 at 41.2 kb. For this germplasm, our marker spacing was approximately 18.1 kb (~ 606 Mb genome size divided by 33,519 markers). For this reason, it seems that we achieved sufficient marker coverage in our germplasm.
Genome-wide association study
The significant threshold for the GWAS analysis was equal to 1.5 × 10−6 (0.05/33,519) following the Bonferroni method. Two hundred and forty-six unique markers showed a total of 1,285 significant marker trait associations with time of leaf budburst and flowering-related phenotypes across all traits and seasons (Supplementary Data 2). These 1,285 associations were clustered in 16 multi-trait QTL distributed on eight different LGs, of which one had unknown genomic positions (Table 2). LG8 had the highest number of QTL (six) followed by LG4 with three different QTL. The R2 values for all the sixteen QTL across different traits and seasons ranged between 0.08 and 0.23 indicating the presence of major QTL controlling different traits (Fig. 3; Table 2; Supplementary Data 2). Within 20 kb flanking the highly associated markers, we detected a number of flowering related domains such as ESCAROLA, FERONIA, TCP20, FERONIA and MYB for all QTL in general (Supplementary Data 2).

Manhattan plots, Quantile–quantile plots and Boxplots from association analysis of three phenological traits; (a) leafing time, (b) female flower abundance, (c) lateral bud flowering.
QTL05 was associated with seven different traits (budburst, leafing, first and last blooming dates for male and female as well as nut setting type) which is the largest number compared to other QTL, with R2 values between 6 and 16%. The allelic effect ranged from 2 to 5 days for the first six traits and 0.18 points on the scale of nut setting type (Supplementary Data 2). There were seven QTL controlling both female abundance and lateral bud flowering distributed on LG4, LG7 and LG8, as well as one QTL with unknown position. The best R2 values for these QTL ranged between 9 and 18% for female abundance and 12–23% for lateral bud flowering and the allelic substitution effect ranged from 0.84 to 1.3, and 17.5–30%; respectively (Supplementary Data 2). One of these QTL, QTL10, was also associated with male flowering period explaining 11% from its variation and having an effect of one day.
QTL02, QTL08 and QTL14 were all associated with first and last male bloom data and inflorescence habit explaning from 6 to 11%, 7–12% and 8–18% for each of the phenotypic variation for the three traits; respectively. The allelic substitution effects ranged from 2.8 to 4.9 days for male blooming and 0.4–1 for inflorescence habit. QTL08 was also associated with first female blooming date with R2 value of 6% and allelic effect of 3.1 days. QTL07 and QTL13 were associated with the first and last blooming dates for male and female (R2 between 6 and 14% and effect between 3.9 and 8.6 days) as well as inflorescence habit with R2 = 20% and effect = 1.3 for QTL07 and R2 = 15% and effect = 1.2 for QTL13. The remaining QTL (QTL01, QTL03 and QTL15) were associated with budburst, leafing and last make blooming dates, explaining from 8 to 12%, 6–11% and 6–8% for each of the three traits; respectively. QTL03 was also associated with first female blooming date while QTL15 was assocaited with first female blooming date. The allelic effects ranged between 2.2 and 5.1 days.
Discussion
Phenotypic data and genetic diversity
Time of leaf budburst and flowering habits are complex phenological traits that are influenced by a variety of physiological and environmental factors and they vary widely among genotypes. Genetic and agronomic improvements have greatly increased the yield potential of plants. Therefore, it is very important to associate these traits genetically and to establish the correlation between them40. In the present study, significant phenotypic variations were detected for 13 important phenological characteristics related to time of leaf budburst and flowering-related traits among walnut accessions. We obtained high narrow-sense heritability values for budburst and leafing dates as well as for the first and the last female and male flowering dates, while the values for female and male flowering periods were low as reported previously14,15. The narrow-sense heritability value for lateral bud flowering (0.538) was intermadiate in thsi study, while Marrano et al.14 obtained very high value (0.98).
In this study, positive correlations were detected between time of budburst and female-male blooming dates with similar findings by Bernard et al.15, while there were no correlations between female and male blooming dates as previously indicated15. Lateral bud flowering is one of yield parameters in walnut, and a negative correlation (− 0.24) was reported14 between leafing date and lateral bud flowering, while a positive correlation (0.39) was found in this study. This can be explained by different populations used in two studies. Long flowering period is an important trait due to get high percentage of nut settings. Male flowering period was usually shorter than female ones in this study as reported previously15. Catkin abundance was found as a strong indicator for long male flowering period due to high correlation between them. In this study, female flowering period and female flower abundance had positive correlations with most of the traits, while male flowering period and catkin abundance had negative ones. Low correlations and low istatistical significances were indicated between female-male flowering period and other traits15.
Choosing the right populations is critical for successful association mapping. The selected natural walnut population must have a sufficiently high genetic diversity. Our walnut accessions consisted of 188 walnut genoypes which are common cultivars in USA and in France as well as some important domestic Turkish walnut cultivars and genotypes that have outstanding characteristics. Therefore, our population had a very wide genetic diversity. Genetic diversity of different J. regia L. germplasm has previously been described in various studies12,13,14,15,16,17,41,42,43,44,45,46,47,48 and it was shown that walnut genotypes conserved a high level of genetic diversity (Fig. 2; Supplementary Fig. S2). The kinship (Supplementary Fig. S2) analyses confirmed the presence of massive diversity in this germplasm. Moreover, LD decayed quickly at around 20 kb indicated large effective population size for the walnut population.
Previous population genetic structure studies in J. regia used SSR markers and focused on germplasms distributed on restricted geographical regions with limited diversity such as Indian genotypes46, trees from cold temperate areas of the USA and Europe44, and Chinese genotypes43,48. Another study investigated the natural population of J. regia in the eastern Italian Alps where moderate genetic diversity was found45. Using the Axiom 700 K SNP array, Arab et al.12 divided a set of 95 Iranian accessions into four subpopulations. The same SNP array was used by Bernard et al.15, and the genotypes originating from Western Europe and America were located in one cluster and the genotypes of Eastern Europe and Asia origin were in the other cluster, while French, American and native walnut genotypes were not sharply separated in this study. Erman et al.16 used a total of 13,611 DArT derived SNP markers, and two peaks were detected at K = 3 and K = 5 in the cluster analysis of 154 walnut accessions, which did not cluster according to their geographical origin. The walnut cultivars and outstanding genotypes were included in this study as the analysis revealed the existence of ten ancestral subpopulations. Even though many genotypes with similar phenotypes were genetically clustered together, the ADMIXTURE analysis at k = 2 showed considerable mixing between early and late blooming accessions.
GWAS
Time of leaf budburst is a very important trait for fruit trees because of the importance of identifying earliness or lateness and mechanisms for their adaptability to different ecological conditions. There have been few association mapping studies on budburst and leafing time for tree species. Flowering-related traits are usually considered as important indicators for productivity. Association mapping studies have been carried out in different plants related to flowering traits such as rice49,50 and apple51. In wheat, barley, and soybean, for example, major genes controlling flowering time were reported to be associated with yield in different climates52,53,54. In walnut, Kefayati et al.55 identified a major QTL for leafing time using a ‘Chandler × Kaplan-86’ F1 segregating population. Similarly, a complex genetic archtictue for flowering related traits in a diverse germplasm was also observed in walnut by Bernard et al.15 with multiple major and minor genes contributing to the variation of different traits. Marrano et al.14 also detected several QTL associated with leafing and harvest dates. In the present study, we detected the first markers associated with some characteristics such as catkin abundace, female flower abundance and nut setting type, which are very important for yield-related traits in walnut. Besides, we revealed that several highly significant markers associated with major multi-trait QTL for different phenological traits. In general and as expected, almost each detected QTL was associated with multiple significantly correlated traits.
A number of the QTL reported in this study overlapped with or were flanked by genes with annotations that are known to play critical roles in flowering time in other plant species. QTL01 is only 3.4 kb away from the gene WALNUT_00020424 which is related to FAR1 (Supplementary Datas 1 and 2). FAR1 is a transcription factor that regulates the phytochrome A signaling pathway in Arabidopsis thaliana and regulates the gene ELF4 that is essential to maintain the circadian rhythm56,57. The most significant SNP associated with QTL08 (100033978|F|0–67:A>G–67:A>G) is located within the gene WALNUT_00018132 that encoded a BEACH domain-containing protein. The BEACH domain was previously reported to be associated with flowering and flower organ development in Arabidopsis58. Multiple markers on the scaffold jcf7180001222160 showed highly significant associations with QTL10. Of those, the markers 12413277 and 23443558 showed highly significant association with male flowering time, female abundance and lateral bud flowering in all seasons. These markers were linked to the genes WALNUT_00006818 and WALNUT_00006828 which encoded DNA-binding protein ESCAROLA and auxin efflux carrier component 8 (PIN8), respectively (Supplementary Data 2). ESCAROLA was previousuly reported to be responsible for modulating hypocotyl growth inhibition in response to light in Arabidopsis59 and affecting leaf senescence60,61 as well as flowering time62. PIN8 was previously reported to be involved in regulating auxin dependent transcriptional activity which has a major effect on the growth of lateral buds63. Thus, it seems that QTL10 is associated with multiple genes that are controlling flowering and leafing traits.
QTL05 is another example of QTL that seems to be associated with multiple genes. This QTL had the largest number of highly significant marker-trait associations that are located on different scaffolds with high LD with each other (Supplementary Data 2). Seven of these markers overlapped with or flanked genes with flowering and leafing related annotations. These annotations included FERONIA, TCP20, auxin-induced, ankyrin and MYB related proteins. FERONIA is known to play a critical role in female control of pollen tube, growth, development, hormon signalling and stress tolerance in different organisms64; while TCP20 was previously reported to modulate flowering65. McClure66 reported an SNP associated with flowering time in the apple that is linked to an ankyrin repeat-containing protein At2g01680-like, which is the same annotation for the walnut gene WALNUT_00004309 that overlapped with the marker 7394079 in this study. Three markers also associated with QTL05 were linked to MYB related genes which are WALNUT_00010727, WALNUT_00028043, and WALNUT_00031245. MYB is a large gene family that has been extensively studied and many members of this family were previously reported to affect anther development, flowering time and to get upregulated in flowers in different plants including Arabidopsis thaliana, rice, and bamboo67,68,69,70. QTL03 was also linked to the MYB related gene WALNUT_00008342 (Supplementary Data 2).
Conclusion
In summary, our study confirmed that time of leaf budburst, leafing time and flowering-related characteristics are complex phenotypic traits. In this study, a large amount of phenotypic variation was observed for the phenological traits studied and presented an important opportunity to examine the genetic architecture of these complex traits. This is the first GWAS study in walnut providing results from high number (13) of traits studied together, and results for the first time from several traits such as catkin and female flower abundance, nut setting type, and inflorescence habit. Using GWAS, we identified a large number of loci and significant 16 QTL associated with leaf budburst time and flowering-related traits. These genes can be used in marker-based breeding programs for earliness/lateness and productivity in the walnut. Moreover, the high resolution of linkage disequilibrium mapping in this study will facilitate the determination of the exact causal variants and underlying genes controlling these traits in future studies. Thus, this study is a step toward understanding the molecular pathway involved in the flowering response in walnut. Future research should focus on the effect of combining different alleles of these genes to characterize their impact on flowering time. Controlling the time of flowering is one of the main factors affecting walnut yield, thus, ensuring flowering at the optimal time will avoid yield loss. The markers detected in this research can facilitate the selection of walnut trees harboring favorable combinations of flowering genes to optimize its timing.
References
Manning, W. E. The classification within the Juglandaceae. Ann. Missouri Bot. Gard. 65(4), 1058–1087 (1978).
Hendricks, L., McGranahan, G., Ramos, D., Iwarkiri, B. & Forde, H. Walnut Orchard Management establishing the walnut orchard Vol. 24410, 46–51 (Division of Agriculture and Natural Resources, California University Publication, Oakland, 1985).
Woodworth, R. H. Meiosis of microsporogenesis in the Juglandaceae. Am. J. Bot. 17(9), 863–869 (1930).
Horjales, M., Redondo, N., Blanco, A. & Rodríguez, M. Nuclear DNA amounts of trees and shrubs. NACC (Nova Acta Científica Compostelana). Bioloxía (España) (2003).
Martínez-García, P. J. et al. The walnut (Juglans regia) genome sequence reveals diversity in genes coding for the biosynthesis of non-structural polyphenols. Plant J. 87, 507–532 (2016).
Bai, W. N. et al. Demographically idiosyncratic responses to climate change and rapid Pleistocene diversification of the walnut genus Juglans (Juglandaceae) revealed by whole-genome sequences. New Phytol. 217, 1726–1736 (2018).
Stevens, K. A. et al. Genomic variation among and within six Juglans species. G3 Genes Genomes Genetics 8, 2153–2165 (2018).
Zhu, T. et al. Sequencing a Juglans regia × J. microcarpa hybrid yields high-quality genome assemblies of parental species. Hortic. Res. 6, 55 (2019).
Marrano, A. et al. High quality chromosome-scale assembly of the walnut (Juglans regia L.) reference genome. bioRxiv https://doi.org/10.1101/809798 (2020).
Faostat. Food and Agricultural Organization of the United Nations, Rome, Italy (2019). https://faostat.fao.org. Accessed 7 September 2019.
You, F. M. et al. Genome-wide SNP discovery in walnut with an AGSNP pipeline updated for SNP discovery in allogamous organisms. BMC Genomics 13, 354 (2012).
Arab, M. M. et al. Genome-wide patterns of population structure and association mapping of nut-related traits in Persian walnut populations from Iran using the Axiom J. regia 700K SNP array. Sci. Rep. 9, 6376 (2019).
Marrano, A. et al. A new genomic tool for walnut (Juglans regia L.): Development and validation of the high-density AxiomTM J. regia 700K SNP genotyping array. Plant Biotechnol. J. 17, 1027–1036 (2019).
Marrano, A. et al. Deciphering of the genetic control of phenology, yield, and pellicle color in Persian walnut (Juglans regia L.). Front. Plant Sci. 10, 1140 (2019).
Bernard, A. et al. Association and linkage mapping to unravel genetic architecture of phenological traits and lateral bearing in Persian walnut (Juglans regia L.). BMC Genomics. 21, 203 (2020).
Erman, E. et al. Association mapping of several nut characters in walnut (Juglans regia L.). Turk. J. Agric. Forest. 44, 208–227 (2020).
Arab, M. M. et al. Combining phenotype, genotype and environment to uncover genetic components underlying water use efficiency in Persian walnut. J. Exp. Bot. 71, 1107–1127 (2020).
Torkamaneh, D. et al. Genome-wide SNP calling from genotyping by sequencing (GBS) data: A comparison of seven pipelines and two sequencing technologies. PloS One 11(8), e0161333 (2016).
Li, H. et al. A high density GBS map of bread wheat and its application for dissecting complex disease resistance traits. BMC Genomics 16(1), 216 (2015).
Brachi, B. et al. Linkage and association mapping of Arabidopsis thaliana flowering time in nature. PLoS Genet. 6(5), e1000940 (2010).
Famula, R. A., Richards, J. H., Famula, T. R. & Neale, D. B. Association genetics of carbon isotope discrimination and leaf morphology in a breeding population of Juglans regia L. Tree Genetics Genomes 15, 6 (2019).
Beauvieux, R., Wenden, B. & Dirlewanger, E. Bud dormancy in perennial fruit tree species: A pivotal role for oxidative cues. Front. Plant Sci. 9, 657 (2018).
Hassankhah, A., Vahdati, K., Rahemi, M. & Sarikhani, S. Persian walnut phenology: Effect of chilling and heat requirements on Budbreak and flowering date. Int. J. Hort. Sci. Tech. 4, 259–271 (2017).
Akca, Y. & Ozongun, S. Selection of late leafing, late flowering, laterally fruitful walnut (Juglans regia) types in Turkey. N. Zeal. J. Crop Hortic. Sci. 32(4), 337–342 (2004).
Germain, E. Inheritance of late leafing and lateral bud fruitfulness in walnut (Juglans regia L.) phenotypic correlations among some traits of the trees. Acta Hortic. 284, 125–134 (1990).
Eskandari, S., Hassani, D. & Abdi, A. Investigation on genetic diversity of Persian walnut and evaluation of promising genotypes. Acta Hortic. 705, 159–166 (2005).
IPGRI. Descriptors for walnut (Juglans spp.). (International Plant Genetic Resources Institute, Rome, 1994).
UPOV. International union for the protection of new varieties of plants. Guidelines for the conduct of tests for distinctness, uniformity and stability. Walnut (Juglans regia L.) 37 (Geneva, 2015).
Doyle, J. & Doyle, J. Isolation of plant DNA from fresh tissue. Focus 12, 13–15 (1990).
Kafkas, S., Ozkan, H. & Sutyemez, M. DNA polymorphism and assessment of genetic relationships in walnut genotypes based on AFLP and SAMPL markers. J. Am. Soc. Hortic. Sci. 130, 585–590 (2005).
Kilian, A. et al. Data Production and Analysis in Population Genomics 67–89 (Springer, New York, 2012).
Hill, W. & Weir, B. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 33, 54–78 (1988).
Alexander, D. H., Novembre, J. & Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 19, 1655–1664 (2009).
Purcell, S. et al. PLINK: A toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 81, 559–575 (2007).
Yang, J. et al. Common SNPs explain a large proportion of the heritability for human height. Nat. Genet. 42, 565 (2010).
Lipka, A. E. et al. GAPIT: Genome association and prediction integrated tool. Bioinformatics 28, 2397–2399 (2012).
VanRaden, P. M. Efficient methods to compute genomic predictions. J. Dairy Sci. 91, 4414–4423 (2008).
Bonferroni, C. E. Teoria statistica delle classi e calcolo delle probabilità Vol. 8, 3–62 (Pubblicazioni del R Istituto Superiore di Scienze Economiche e Commerciali di Firenze, Rome, 1936).
Kefayati, S. Cevizin SSR, SNP ve DArT markörleri ile yoğun genetik haritasının oluşturulması PhD thesis, University of Çukurova, (2018).
Farneti, B. et al. Genome-wide association study unravels the genetic control of the apple volatilome and its interplay with fruit texture. J. Exp. Bot. 68, 1467–1478 (2017).
Doğan, Y., Kafkas, S., Sütyemez, M., Akça, Y. & Türemiş, N. Assessment and characterization of genetic relationships of walnut (Juglans regia L.) genotypes by three types of molecular markers. Sci. Hortic. 168, 81–87 (2014).
Pollegioni, P. et al. Landscape genetics of Persian walnut (Juglans regia L.) across its Asian range. Tree Genetics Genomes. 10, 1027–1043 (2014).
Wang, H., Wu, W., Pan, G. & Pei, D. Analysis of genetic diversity and relationships among 86 Persian walnut (Juglans regia L.) genotypes in Tibet using morphological traits and SSR markers. J. Hortic. Sci. Biotechnol. 90, 563–570 (2015).
Ebrahimi, A., Zarei, A., McKenna, J. R., Bujdoso, G. & Woeste, K. E. Genetic diversity of Persian walnut (Juglans regia) in the cold-temperate zone of the United States and Europe. Sci. Hortic. 220, 36–41 (2017).
Vischi, M. et al. Genetic diversity of walnut (Juglans regia L.) in the Eastern Italian Alps. Forests 8, 81 (2017).
Shah, U. N., Mir, J., Ahmed, N. & Fazili, K. M. Assessment of germplasm diversity and genetic relationships among walnut (Juglans regia L.) genotypes through microsatellite markers. J. Saudi Society Agric. Sci. 17, 339–350 (2018).
Bernard, A., Barreneche, T., Lheureux, F. & Dirlewanger, E. Analysis of genetic diversity and structure in a worldwide walnut (Juglans regia L.) germplasm using SSR markers. PloS One 13(11), e0208021 (2018).
Han, H. et al. Genetic diversity and population structure of common walnut (Juglans regia) in China based on EST-SSRs and the nuclear gene phenylalanine ammonia-lyase (PAL). Tree Genetics Genomes 12, 111 (2016).
Ordonez, S. A. Jr., Silva, J. & Oard, J. H. Association mapping of grain quality and flowering time in elite japonica rice germplasm. J. Cereal Sci. 51, 337–343 (2010).
Huang, X. et al. Genome-wide association study of flowering time and grain yield traits in a worldwide collection of rice germplasm. Nat. Genet. 44, 32 (2012).
Urrestarazu, J. et al. Genome-wide association mapping of flowering and ripening periods in apple. Front. Plant Sci. 8, 1923 (2017).
Zhang, J. et al. Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genomics 16, 217 (2015).
Al-Abdallat, A. et al. Assessment of genetic diversity and yield performance in Jordanian barley (Hordeum vulgare L.) landraces grown under Rainfed conditions. BMC Plant Biol. 17, 191 (2017).
Ogbonnaya, F. C. et al. Genome-wide association study for agronomic and physiological traits in spring wheat evaluated in a range of heat prone environments. Theoret. Appl. Genetics 130, 1819–1835 (2017).
Kefayati, S. et al. First simple sequence repeat-based genetic linkage map reveals a major QTL for leafing time in walnut (Juglans regia L.). Tree Genetics Genomes 15, 13 (2019).
Li, G. et al. Coordinated transcriptional regulation underlying the circadian clock in Arabidopsis. Nat. Cell Biol. 13, 616 (2011).
Johansson, M. & Staiger, D. Time to flower: Interplay between photoperiod and the circadian clock. J. Exp. Bot. 66, 719–730 (2014).
Van Nocker, S. & Ludwig, P. The WD-repeat protein superfamily in Arabidopsis: Conservation and divergence in structure and function. BMC Genomics 4, 50 (2003).
Street, I. H., Shah, P. K., Smith, A. M., Avery, N. & Neff, M. M. The AT-hook-containing proteins SOB3/AHL29 and ESC/AHL27 are negative modulators of hypocotyl growth in Arabidopsis. Plant J. 54, 1–14 (2008).
Lim, P. O., Kim, H. J. & Gil Nam, H. Leaf senescence. Annu. Rev. Plant Biol. 58, 115–136 (2007).
Lim, P. O. et al. Overexpression of a chromatin architecture-controlling AT-hook protein extends leaf longevity and increases the post-harvest storage life of plants. Plant J. 52, 1140–1153 (2007).
Weigel, D. et al. Activation tagging in Arabidopsis. Plant Physiol. 122, 1003–1014 (2000).
Bosco, C. D. et al. The endoplasmic reticulum localized PIN8 is a pollen-specific auxin carrier involved in intracellular auxin homeostasis. Plant J. 71, 860–870 (2012).
Duan, Q. & Cheung, A. Y. Context-specific dependence on FERONIA kinase activity. FEBS Lett. 592, 2392–2394 (2018).
Lucero, L. E., Manavella, P. A., Gras, D. E., Ariel, F. D. & Gonzalez, D. H. Class I and class II TCP transcription factors modulate SOC1-dependent flowering at multiple levels. Molecular Plant 10, 1571–1574 (2017).
McClure, K. Exploring horticulturally important traits in apple using genetic mapping in populations of differing size and composition. Ph.D. Thesis, The University of Guelph, Department of Plant Agriculture 213 (2017).
Kuno, N. et al. The novel MYB protein EARLY-PHYTOCHROME-RESPONSIVE1 is a component of a slave circadian oscillator in Arabidopsis. Plant Cell 15, 2476–2488 (2003).
Aya, K. et al. Gibberellin modulates anther development in rice via the transcriptional regulation of GAMYB. Plant Cell 21, 1453–1472 (2009).
Zhang, L. et al. The wheat MYB-related transcription factor TaMYB72 promotes flowering in rice. J. Integr. Plant Biol. 58, 701–704 (2016).
Ge, W. et al. Main regulatory pathways, key genes and micro RNA s involved in flower formation and development of moso bamboo (Phyllostachys edulis). Plant Biotechnol. J. 15, 82–96 (2017).
Acknowledgements
The authors would like to thank Dr. Hans D. Daetwyler (Agriculture Victoria) for reviewing the manuscript.
Funding
This work was supported by Scientific Research Projects Units of Kahramanmaraş Sütçü İmam University (Project No. 2018/3-31M) and Cukurova University (Project No. FBA-2018-10054).
Author information
Authors and Affiliations
Contributions
Sa.K. M.S. Ş.B.B and A.J. planned and designed the experiment. Ş.B.B. Sa.K. A.P. M.S. and Si.K. conducted the phenotyping and genotyping. A.J. Ş.B.B. and Sa.K. analyzed the data. Ş.B.B., A.J., M.S., and Sa.K. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bükücü, Ş.B., Sütyemez, M., Kefayati, S. et al. Major QTL with pleiotropic effects controlling time of leaf budburst and flowering-related traits in walnut (Juglans regia L.). Sci Rep 10, 15207 (2020). https://doi.org/10.1038/s41598-020-71809-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-71809-x
This article is cited by
-
Selecting the superior late-leafing genotypes of Persian walnut (Juglans regia L.) using morphological and pomological evaluations
BMC Plant Biology (2023)
-
Genome-wide characterization and development of SSR markers for genetic diversity analysis in northwestern Himalayas Walnut (Juglans regia L.)
3 Biotech (2023)
-
Morphological to Molecular Markers: Plant Genetic Diversity Studies in Walnut (Juglans regia L.)—A Review
Erwerbs-Obstbau (2023)
-
Genome-wide association analysis of 101 accessions dissects the genetic basis of shell thickness for genetic improvement in Persian walnut (Juglans regia L.)
BMC Plant Biology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
