
Abstract
Fibrillin-1 (FBN1) is responsible for haploinsufficient and autosomal dominant Marfan syndrome. Even in the same Marfan pedigree, penetrance and expressivity in heterozygous individuals can differ and result in variable disease onset and severity. Thus, other factors in addition to mutations in FBN1 are likely to contribute to the disease. In this study, we examined the regulation of FBN1 in porcine Marfan syndrome model, focusing on DNA methylation patterns distinguishable as wild-type (WT) and FBN1 null (KO) alleles in heterozygous cells. Most importantly, the ratio of the transcriptionally active hypomethylated WT allele was altered during cellular passage and highly correlated with FBN1 mRNA level compared with that in the KO allele. Transcribed FBN1 RNA from the KO allele was abolished after splicing coupled with translational initiation, suggesting that the functional FBN1 mRNA levels were affected by DNA methylation of the WT allele.
Similar content being viewed by others
Introduction
Fibrillin-1 (FBN1) encodes the microfibril protein, fibrillin, which is expressed in various connective tissues and is a responsible gene for Marfan syndrome1,2. Marfan syndrome is caused by the inheritance of one mutated null allele and classified as autosomal dominant3. However, Marfan patients exhibit phenotypic variations even in the same pedigree with an identical mutated null allele1,4. In addition, patients with low levels of the FBN1 expression suffer an increased risk of ectopia lentis, pectus excavatum, and aorta abnormalities5. Therefore, the expression level of FBN1, which is transcribed and translated into a functional fibrillin protein, is associated with disease onset and the severity of Marfan syndrome.
One cause of Marfan syndrome is haploinsufficiency, a condition with one mutated null allele and one normal allele. Haploinsufficiency is induced by a decrease in the amount of functional protein to less than half that produced under diploid expression6. Our Marfan model pigs7 exhibit haploinsufficiency with phenotypic variations in disease onset, mimicking the condition in humans, wherein differences in disease phenotypes or affected tissues have been reported even among patients from the same pedigree. In contrast, inheritance of two null mutant alleles is sufficient to cause autosomal recessive diseases. Thus, unlike autosomal recessive diseases, Marfan syndrome caused by autosomal dominant haploinsufficiency are unlikely explained completely by genetic mutations, and the molecular mechanism(s) that cause such phenotypic variation must be elucidated.
DNA methylation in transcriptional regulatory regions is crucial for tissue/cell type-specific gene expression8,9,10 and plays an important role in cellular differentiation and mammalian development11,12,13. Sodium bisulfite sequencing is a conventional method for DNA methylation analysis and identifies DNA methylation patterns in each sequenced fragment that can be recognized as one allele (cell) in a tissue or cell population14,15. Based on this concept, we previously focused on transcriptionally active reduced methylation allele (RM allele), which was described as DNA hypomethylated alleles (Hypo-alleles) in our previous study14,15 that can be recognized by the high ratio of unmethylated CpGs in each sequenced fragment (allele), and the RM allele ratio of a non-imprinted autosomal gene can be used to estimate the proportion of the cell type expressing that gene in the tissue or cell population14,15. Therefore, the RM allele model can be applied to detect transcriptional regulation of each allele in diploid cells in certain cell populations.
In human Marfan patients, an identical FBN1 null mutant allele is inherited in a pedigree and can present different disease onset and severity among individuals in the same family. In humans, genetic sequences differ among individuals in the same pedigree, making it impossible to eliminate influences of genetic differences other than that of the gene responsible for the disease on the variations of disease phenotypes in affected individuals. This hinders the ability to elucidate the cause of phenotypic variations of genetic diseases. At present, differences in nucleotide sequences of genetic modifiers are thought to be responsible for phenotypic variations such as varied disease onset and/or severity in genetic diseases. In contrast, we produced FBN1 heterozygous knockout (het KO) cloned pigs using genome editing and somatic cell nuclear transfer as an animal model for human Marfan syndrome7. Previous studies on Marfan patients have reported two types of FBN1 genetic mutations, haploinsufficiency and dominant-negative16. Although both types of FBN1 mutations are important for understanding the pathogenic mechanisms of Marfan syndrome, disease onset of our FBN1 KO cloned pig model was caused by haploinsufficiency. This was confirmed by the absence of mutant mRNA with a premature stop codon7. These Marfan model pigs exhibit a broad spectrum of abnormal phenotypes that are also found in humans7. The FBN1 het KO cloned pigs each have a syngeneic background because of their production by somatic cell nuclear transfer; however, these Marfan model cloned pigs exhibit a wide range of disease phenotypes including non-penetrance. Thus, the disease onset and severity of the Marfan model cloned pigs most likely result solely from a genetic mutation by a 1-bp deletion within the FBN1 coding region that causes a frameshift and creates a premature stop codon7 and not from genomic sequence differences outside FBN1.
In our previous study, we reported that the porcine FBN1 is regulated by DNA methylation, and FBN1 mRNA levels are associated with RM allele ratios of an FBN1 upstream region of the CpG island shore17. We also found that the nucleotide sequence of the CpG island shore of porcine FBN1 is more highly homologous to human FBN1, including the number of methylatable CpG dinucleotides than the mouse Fbn1. These results together with the results of FBN1 het KO cloned pigs suggest the possibility that FBN1 expression is regulated by variable changes in epigenetic modification in Marfan patients. To further investigate the epigenetic regulation of FBN1 in cells with one mutated FBN1-null allele, which is similar to human Marfan syndrome, an experimental tool for allele-distinguished DNA methylation analysis was designed by producing FBN1 het KO pigs in which the FBN1 CpG island shores of the WT and KO alleles have a single nucleotide polymorphism (SNP). Here, we analyzed primary fibroblast cells and adult tissues derived from FBN1 het KO pigs with the allele-distinguishable SNP in the CpG island shore and focused on RM allele ratios within the FBN1 CpG island shore, especially in the WT allele.
Results
Establishment of primary fibroblast cell lines derived from FBN1 het KO pigs with a SNP that differentiates normal (WT) and FBN1 null (KO) alleles
In an FBN1 homozygous KO pig produced by crossing FBN1 het KO pigs7, no FBN1 mRNA was detected by RT-PCR and real-time RT-PCR (Supplementary Fig. 1a,b). However, an FBN1 preRNA that contained exons and introns before splicing was clearly detected in the FBN1 homozygous KO cells and WT cells (Supplementary Fig. 1a), indicating that FBN1 is transcribed from both normal (WT) and FBN1-mutated null (KO) alleles, while the spliced and mature FBN1 mRNA detected by RT-PCR analysis was the transcript only from the WT allele. Thus, RNA from the KO allele apparently existed before splicing, whereas spliced mature KO mRNA became degraded by a nonsense-mediated RNA decay mechanism that recognizes the premature stop codon created by the 1-bp deletion within the FBN1 coding region of the KO allele. This also indicates that, at least in our Marfan model pigs, dominant negative effects of the KO allele are not likely to be responsible for disease onset and severity due to absence of mature mRNA. In addition, treatment of the FBN1 het KO fibroblast cells (KO-239-2 and KO-239-4) with 5-aza-2′-deoxycytidine (5-aza-dC), an inhibitor of DNA methyltransferase, caused a significant increase in the amount of FBN1 mRNA (Supplementary Fig. 2a). These results suggest that transcription of FBN1 mRNA from the WT allele is controlled by DNA methylation. Furthermore, our promoter assay combined with in vitro DNA methylation of the FBN1 CpG island shore revealed that promoter activity was clearly reduced by DNA methylation of the FBN1 CpG island shore (Supplementary Fig. 2b). These results indicate that FBN1 is regulated, at least in part, by DNA methylation of its CpG island shore.
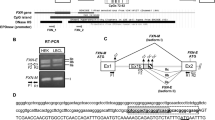
In order to investigate the regulation of FBN1 mRNA level in porcine FBN1 het KO cells, we attempted to analyze DNA methylation status of the FBN1 CpG island shores of WT and KO alleles, independently. As a tool to perform allele-distinguished DNA methylation analysis of the FBN1 CpG island shore, we first produced FBN1 het KO pigs that contained an allele-distinguishable single nucleotide polymorphism (SNP) within the FBN1 CpG island shore. Comparison of the genomic sequences of the FBN1 CpG island shore in WT and FBN1 het KO pigs could successfully locate a SNP that is a cytosine in both alleles of WT pigs and a thymine in both alleles of the FBN1 het KO pigs. We produced F1 FBN1 het KO pigs that had both cytosine and thymine SNPs that corresponded to WT and KO alleles, respectively (Fig. 1a). Using neonatal tail-cartilage from SNP-containing FBN1 het KO pigs, we established primary fibroblast cell lines from six F1 het KO pigs (KO-239-2, KO-239-3, KO-239-4, KO-239-5, KO-241-2, and KO-241-5) as well as fibroblast cell lines from two F1 WT littermates (WT-239-1 and WT-241-1). Analysis of FBN1 expression in these fibroblast cell lines by RT-PCR revealed that the amount of the FBN1 mRNA was significantly lower in the FBN1 het KO cells than in WT cells (Fig. 1b). On the other hand, no significant difference was observed in the amount of FBN1 preRNA before splicing between FBN1 het KO and WT cells, which was as expected from the results of homozygous KO pigs (Supplementary Fig. 1a).

FBN1 mRNA and preRNA levels in porcine FBN1 het KO fibroblast cells depending on the normal (WT) and FBN1-mutated null (KO) alleles. (a) The upper panel shows a schematic of SNP and RT-PCR primer positions in the porcine FBN1 locus. The RT-PCR primers were designed to amplify regions including the 1-bp deletion site (guanine, G) in the FBN1 exon 10. FBN1 mRNA and preRNA were amplified with the specific primer sets (F1 and R1) and (F3 and R3), respectively. White and black squares indicate the FBN1 CpG island and the CpG island shore, respectively. The lower panel shows sequencing data of the SNP position in the FBN1 CpG island shore. Open triangles indicate the position of the SNP-identified top strand in the FBN1 het KO pigs (C: WT allele, T: KO allele). The same position of the SNP in the complementary strand is G in the WT allele and A in the KO allele, and was amplified by sodium bisulfite PCR. (b) Expression levels of the FBN1 mRNA and preRNA in wild-type fibroblast cells (n = 3) and FBN1 het KO fibroblast cells (n = 6) were evaluated by RT-PCR. (c) Expression levels of the FBN1 mRNA, preRNA, and ratio of mRNA to preRNA (mRNA/preRNA) in FBN1 het KO fibroblast cells (n = 6) collected at passages 6 and 20 (P6 and P20) were measured by RT-PCR. (d) Changes of FBN1 mRNA levels from P6 to P20 in each FBN1 het KO fibroblast cell line (n = 6). These data are based on the FBN1 mRNA data in (c). The relative expression levels of FBN1 mRNA and preRNA in (b,c) were normalized to GAPDH expression and shown as mean ± SD (n = 3). Statistical comparisons of the expression levels were performed using the Student’s t-test, and statistical significance was set as p < 0.05.
Previous studies have reported that differences in primary cultured cell passage number can affect gene expression18,19,20. In addition, disease onset tends to vary among Marfan’s patients even in the same pedigree. To investigate the regulation of FBN1 expression in the in vitro het KO cell cultures, we evaluated on the passage number of FBN1-expressing fibroblast cells by RT-PCR. Differences in FBN1 het KO cell passage number revealed that the average amounts of both FBN1 mRNA and preRNA of the six FBN1 het KO fibroblast cell lines were comparable between P6 (early passage) and P20 (late passage) (Fig. 1c). However, the ratio of FBN1 mRNA to preRNA (mRNA/preRNA) in each cell line tended to decrease at P20 compared with P6. In addition, the FBN1 mRNA level in each het KO line changed with the passage number, and expression patterns in P6 to P20 differed depending on cell line (Fig. 1d). The FBN1 mRNA was only detected from the WT allele by RT-PCR (Supplementary Fig. 1), suggesting that transcription levels of the WT allele changed with the passage number, resulting in variable mature mRNA levels in FBN1-expressing fibroblast cell populations.
Allele-distinguished DNA methylation analysis within the FBN1 CpG island shore in FBN1 het KO fibroblast cells
To investigate the changes of FBN1 mRNA expression with passage number in FBN1 het KO cells, we first focused on DNA methylation status within the FBN1 CpG island shore of each allele with sodium bisulfite sequencing (Fig. 2). The SNP that we found in the CpG island shore was a C/T SNP difference on one strand between the WT and KO alleles. However, bisulfite modification eliminates the information of the C/T SNP because unmethylated cytosines are converted to uracil and then thymine during PCR amplification. Thus, allele-distinguishable bisulfite sequencing was performed using a primer set that could amplify the opposite strand containing G/A SNPs to distinguish the WT (G) and KO (A) alleles (Fig. 1a). Bisulfite PCR uses only for one strand of the double-stranded genomic DNA; the two strands are initially complementary to each other but become non-complementary with bisulfite treatment. Based on the SNPs present on the opposite strand, the G/A SNP is not affected by bisulfite treatment and retains the SNP for allele identification. We used the SNP within the FBN1 CpG island shore (i.e. G: WT allele, A: KO allele) to analyze the DNA methylation status of six FBN1 het KO cell lines at P6 and P20. Based on the methylation level, we previously divided the FBN1 CpG island shore into three sub-regions: the constitutively hypermethylated outside region (O), the constitutively hypomethylated CpG island region (I), and the intermediately and differentially methylated core region of the CpG island shore (S) between O and I17. Focusing on the hypomethylated allele ratio rather than a conventional DNA methylation rate of the examined region, the proportion of a certain cell type can be more precisely estimated in heterogeneous cell populations such as tissues15. In the present study, either hypermethylated or hypomethylated patterns were observed in the S region of the sequenced fragments in FBN1 het KO cells (Fig. 2). Thus, we calculated the RM allele ratio of the WT and KO alleles based on the methylation information of nine CpG sites contained in the S region that is strongly associated with FBN1 expression (Supplementary Fig. 2b), and a fragment with seven or more unmethylated CpGs was defined as an RM allele in accordance with the criteria used in our previous study15. Furthermore, FBN1 het KO cells possess two FBN1 alleles, of which one is normal (WT) and the other is mutated (KO) allele. In other words, one FBN1 het KO cell contains one WT allele, whose hypomethylation results in the expression of functional FBN1 mRNA. Thus, under the experimental condition that bisulfite PCR bears no PCR bias as confirmed in our previous study15, it should be emphasized here that the calculation of the RM allele ratio that focuses only on the WT allele is equivalent to the estimation of the proportion of the cells that contain the hypomethylated and FBN1-producing WT allele in the cultured FBN1 het KO cell population. Taking advantage of this correspondence, we determined the ratio of the cells that were hypomethylated in the CpG island shore of the WT allele, which likely resulted in sufficient expression of functional WT mRNA in the primary fibroblast cell lines.

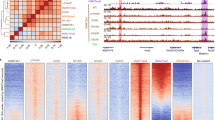
DNA methylation analysis of FBN1 CpG island shores of WT and KO alleles in FBN1 het KO fibroblast cells. A diagram of the porcine FBN1 upstream region is illustrated in the upper panel. The SNP between the WT and KO alleles identified in this study is located in the FBN1 CpG island shore. Sequenced sodium bisulfite PCR fragments including the FBN1 CpG island shore were divided into three sub-regions based on methylation status (O, outside the FBN1 CpG island shore. S, shore region. I, CpG island.). The DNA methylation status of each allele in FBN1 het KO fibroblast cells is represented in the lower panel. Depending on the SNP (G or A) present in the sodium bisulfite PCR fragments, sequenced reads were defined as WT (G) or KO (A) alleles for each cell line collected at P6 (early passage) and P20 (late passage). Horizontal and vertical axes indicate the position of the CpG sites (O, 2 CpGs; S, 9 CpGs; I, 13 CpGs) and the number of sequenced reads (29 to 45 reads), respectively. White and black bars indicate unmethylated and methylated CpGs, respectively, consistent with our previous study15. CpGs within the two dotted lines are in the shore region (S) and whose RM allele ratio was calculated in the present study.
Based on the sodium bisulfite sequencing data (Fig. 2), the RM allele ratio of six FBN1 het KO cell lines generally decreased in P20 compared with P6 in both WT and KO alleles (Fig. 3a), suggesting that changes in DNA methylation status occurred during cell proliferation and passage. In addition, RM allele ratios of both WT and KO alleles decreased from P6 to P20 in three FBN1 KO cell lines (KO-239-2, KO-239-4, and KO-241-5; Fig. 3b). This decrease in the RM allele ratio indicated de novo DNA methylation resulting in DNA hypermethylation within the CpG island shore region during in vitro culture of the fibroblast cells. In particular, in the KO-239-2 line, the RM allele ratio of the KO allele dramatically decreased from P6 to P20 compared with that of the WT allele. On the other hand, RM allele ratios of the WT and KO alleles changed differently in the other three cell lines (KO-239-3, KO-239-5, and KO-241-2) (Fig. 3b). To exclude the possibility that the single nucleotide difference at the SNP site affected DNA methylation, the same analysis was performed using littermate WT pig-derived fibroblast cells and showed that the direction of the changes in the RM allele ratio from P6 to P20 were identical between the two WT alleles (Supplementary Fig. 3). Among the FBN1 het KO cell lines, the variance in the RM allele ratio of the WT and KO alleles were larger in the KO allele than the WT allele (Supplementary Fig. 4), suggesting that in the FBN1 het KO cells the regulation of FBN1 expression by DNA methylation differed between the WT and KO alleles. Overall, four out of the six FBN1 het KO cell lines exhibited the same changes in FBN1 mRNA level and RM allele ratio between P6 and P20 based on the SNP in the WT allele (Fig. 3c). Considering that each het KO cell has one WT allele, the decrease in WT RM allele ratios in these four het KO cell lines likely indicate that the proportion of the cells that can transcribe functional FBN1 mRNA have decreased over time. By plotting the changes in FBN1 mRNA and the RM allele ratio from P6 to P20 for each cell line, a higher correlation was observed for the WT allele than the KO allele, which was expected from their WT RM allele ratios (Fig. 3d). Since the KO allele-derived transcripts are degraded after splicing (Supplementary Fig. 1) and FBN1 expression is regulated by DNA methylation (Supplementary Fig. 2), the functional FBN1 mRNA levels are controlled by DNA methylation within the CpG island shore of the WT allele alone in FBN1 het KO fibroblast cells. Furthermore, FBN1 het KO fibroblast cells (KO-239-2 and KO-239-4) were easier to detach from culture dishes with trypsin/EDTA treatment in vitro than WT fibroblast cells (WT-239-1) (Supplementary Fig. 5a), and the amount of FBN1 mRNA in detached het KO fibroblast cells was significantly lower than that in WT cultures (Supplementary Fig. 5b). Since the microfibril protein, fibrillin, has an important role in the formation of extracellular matrix and cell adhesion21,22, a varied adhesion phenotype among fibroblast cell lines reflects the amount of functional FBN1 mRNA transcribed from the WT allele.

Stochastic DNA methylation changes on both the WT and KO alleles after passage of FBN1 het KO fibroblast cells. (a) RM allele ratio of the FBN1 het KO fibroblast cells (n = 6) at P6 and P20. In the S region, sequenced reads with ≥75% unmethylated CpG sites (i.e. ≥7 out of nine CpGs) were defined as RM alleles. Based on the sodium bisulfite sequencing data in Fig. 2, RM allele ratios were calculated for the WT and KO alleles, respectively, and are shown as average ± SD. Statistical comparisons of the RM allele ratios were performed using the Student’s t-test, and statistical significance was set as p < 0.05. (b) Changes in RM allele ratios of FBN1 het KO fibroblast cells from P6 to P20. Solid and dotted lines indicate the RM allele ratio of WT and KO alleles, respectively. (c) Summary of changes in RM allele ratios and FBN1 mRNA levels in FBN1 het KO fibroblast cells. The data of the FBN1 mRNA is presented in Fig. 1. Black and white triangles indicate the increase and decrease in RM allele ratios and FBN1 mRNA levels from P6 to P20, respectively. (d) Correlation between FBN1 mRNA level and RM allele ratio. The difference in FBN1 mRNA level and RM allele ratio between P6 and P20. Closed circles represent the data of each FBN1 het KO fibroblast cell (n = 6).
Regulation of FBN1 mRNA by DNA methylation of the WT allele in ear cartilage of FBN1 het KO pigs
Next, we attempted to analyze whether FBN1 mRNA transcription is regulated by DNA methylation of the WT allele in vivo using sodium bisulfite sequencing and RT-PCR of porcine tissue to distinguish the WT and KO allele SNPs within the FBN1 CpG island shore and the 1-bp deletion site in FBN1 transcript. It has been reported that FBN1 is highly expressed in a cartilage tissue21 and our data also indicated that FBN1 mRNA was clearly detected in ear cartilage of FBN1 het KO pigs, KO-239-4 and KO-239-2 (Fig. 4a). Differences in the FBN1 expression level in ear cartilage was observed between the two FBN1 het KO pigs analyzed and the FBN1 mRNA level of KO-239-4 tended to be lower than that of KO-239-2. This lower FBN1 mRNA level was also observed in fibroblast cells derived from the same pigs (data not shown). In addition, although the FBN1 preRNA level in ear cartilage was similar between the two FBN1 het KO pigs, a lower mRNA to preRNA ratio was observed in KO-239-4 than in KO-239-2 (Fig. 4a). Analysis of the allele-dependent DNA methylation status of the FBN1 CpG island shore (Fig. 4b) and the FBN1 mRNA level in ear cartilage revealed a higher correlation in the WT allele than in the KO allele (Fig. 4c). These results indicate that functional FBN1 mRNA levels are controlled by DNA methylation on the WT allele in FBN1-expressing tissues of the het KO pigs, and is probably a variable outcome of changes in DNA methylation during in vivo ontogenetic growth and/or aging. Similarly, the ratio of cells representing the DNA hypomethylated state in the CpG island shore of the WT allele is most likely reflected in the total amount of the functional FBN1 mRNA in the tissue.

Allele-distinguished DNA methylation analysis of ear cartilage of the FBN1 het KO pigs. (a) Expression levels of the FBN1 mRNA and preRNA in the ear cartilage of FBN1 het KO (KO-239-4, KO-239-2) and WT (WT-239-1) pigs was measured by RT-PCR. The relative expression levels were normalized to GAPDH expression. The mRNA/preRNA data indicate the ratio of FBN1 mRNA in all transcribed FBN1 RNA. The expression levels are shown as mean ± SD (n = 3). Statistical comparisons of the expression levels were performed using the Student’s t-test, and statistical significance was set as p < 0.05. (b) Allele-distinguished DNA methylation analysis of the same ear cartilage samples from FBN1 het KO and WT pigs was conducted by sodium bisulfite sequencing. Depending on the SNP in the FBN1 CpG island shore, the WT (G) or KO (A) alleles present in the FBN1 het KO pigs were determined in each sequenced read. Although the WT pig analyzed has two WT alleles, one is derived from a WT pig with two G alleles and the other is derived from a WT allele from the FBN1 het KO pig (A). O: hypermethylated outside region. S: variably methylated shore region. I: hypomethylated CpG island. (c) Correlation between FBN1 mRNA level and the RM allele ratio of the S region. Black, gray, and white circles indicate ear-cartilage data from KO-239-4, KO-239-2, and WT-239-1, respectively. In the WT-239-1, the RM allele ratio was calculated using all the sequenced clones of both WT-derived and het-KO-derived FBN1 CpG island shores and was used in both WT- and KO-allele plots without distinguishing the two alleles because two WT alleles were not determined for the WT-239-1 pig in the RT-PCR analysis.
DNA methylation status in the upstream region of FBN1 in human somatic cells
We previously reported that the FBN1 CpG island shore is highly conserved in pig and human genomes17. To investigate whether epigenetic regulation of FBN1 observed in porcine fibroblast cells is conserved in human cells, we analyzed the DNA methylation status of the upstream region of FBN1 in five human somatic cell lines (MRC5, AM936EP, Edom22, PAE551, and Ute1104). In the Infinium Human Methylation450K BeadChip, three probes (probe I, II, and III) were located upstream of the human FBN1; probe I and III were hyper- and hypo-methylated, respectively, in all five human cell lines (Fig. 5a). However, probe II is located within the FBN1 CpG island shore of the human FBN1 and had variable and moderate DNA methylation levels (18.8 to 69.9%) that depended on the cell line.

DNA methylation status of FBN1 CpG island shore and FBN1 expression in human cells. The DNA methylation status of five human somatic cell lines (MRC5, AM936EP, Edom22, PAE551, and UtE1104) (a) and twelve human iPS cell lines (b) were analyzed with the Infinium Human Methylation450K BeadChip. In the human FBN1 upstream region, three probes (probe I, II, and III) that are located in the outside region (probe I), the CpG island shore (probe II), and inside the CpG island (probe III) of the human FBN1 locus. DNA methylation levels are presented as a pie chart; black and white areas indicate the percentages of methylated and unmethylated CpGs, respectively. (c) The correlation between FBN1 mRNA level and DNA methylation level of FBN1 CpG island shore (probe II) was calculated for human iPS cell lines (n = 12). FBN1 expression and DNA methylation data were based on the gene expression array and the Infinium Human Methylation450K BeadChip, respectively.
Next, the correlation between DNA methylation and FBN1 expression in human somatic cells was analyzed. However, the human somatic cells examined exhibited high levels of FBN1 expression; therefore, these data were not suitable for verifying the correlation between DNA methylation and FBN1 expression. In our previous study, human induced pluripotent stem (iPS) cells were shown to differ greatly in DNA methylation status depending on the cell line23. Because of this, further analysis was performed using human iPS cells. DNA methylation analysis using the Infinium Human Methylation450K BeadChip revealed that human iPS cells also exhibited moderate DNA methylation levels of probe II, which is located in the FBN1 CpG island shore. As expected, these methylation levels varied based on the cell lines examined (Fig. 5b). For the human iPS cells, the DNA methylation level of the FBN1 CpG island shore and the FBN1 mRNA level (analyzed using a gene expression array) showed a significantly negative correlation (Fig. 5c). In the porcine samples (Figs. 3d and 4c), a positive correlation was observed when plotting the percentage of alleles where the gene was activated (RM allele) and the level of FBN1 mRNA expression. Conversely, in human iPS cells (Fig. 5c) a negative correlation was observed between the DNA methylation level (an indicator of cell ratio when the gene is in a suppressed state) and FBN1 mRNA expression. Therefore, both in pigs and humans, DNA methylation in the CpG island shore plays a role in the suppression of FBN1 mRNA expression. These results are consistent with data of porcine fibroblast cells and suggest that the human FBN1 is also likely to be regulated by DNA methylation of the FBN1 CpG island shore.
Discussion
In the present study, we investigated how DNA methylation status within the FBN1 CpG island shore of the WT and KO alleles regulates FBN1 expression using allele-specific SNPs in FBN1 het KO fibroblast cells as the tool. Expression levels of FBN1 mRNA differed among fibroblast cell lines and in ear cartilage from littermate het KO pigs. These differences in FBN1 mRNA levels were also observed in human Marfan patients5. In addition, RM allele ratios differed among FBN1 het KO samples, with the WT allele exhibiting a higher correlation with FBN1 mRNA level than the KO allele. A combination of varied DNA methylation of the WT-allele and degradation of KO-allele-derived preRNA suggests that the functional FBN1 mRNA level in cell populations or tissues of het KO pigs is mainly regulated by DNA methylation of the WT allele. Thus, it is becoming more important to investigate epigenetic regulation of WT alleles in Marfan patients with null KO allele and its animal model of our FBN1 het KO pigs.
We produced FBN1 het KO pigs, in which WT and KO alleles could be distinguished using a SNP in the FBN1 CpG island shore. This allowed us to distinguish between WT and KO alleles, and calculate the RM allele ratio of the WT allele that directly represents the proportion of the cells with hypomethylated CpG island shores in the WT allele. The cells with the hypomethylated WT allele most likely express a sufficient level of WT FBN1 mRNA in a certain cell population or tissue (Fig. 6). In contrast, the DNA methylation status (RM allele ratio) of the KO allele should not affect functional mRNA levels because a premature stop codon results in nonsense-mediated RNA decay even if transcript is produced from the KO allele. However, this relation between cell proportion and gene expression level cannot be applied to samples without allele-distinguishable SNPs in the differentially DNA-methylated CpG island shore of het KO cells. Thus, as a practical reason for analyzing epigenetic changes on the WT allele in addition to modeling haploinsufficiency diseases, our FBN1 het KO fibroblast cells distinguishable of the WT and KO alleles are useful for understanding the molecular mechanisms of stochastic downregulation of the responsible gene in relation to epigenetic ambiguity.

A model for FBN1 gene regulation by varied DNA methylation of the CpG island shore of FBN1 het KO cells/tissues. In the FBN1 het KO cells and tissues, the amount of functional FBN1 mRNA is regulated by DNA methylation status within the FBN1 CpG island shore of the WT allele. The null KO allele does not contribute any FBN1 function because the FBN1 preRNA includes a premature stop codon and degraded after splicing coupled with translational initiation. The WT allele is hypomethylated in het KO cells and therefore, the total amount of the functional FBN1 mRNA is sufficient in tissue (cell population), regardless of the methylation status of the KO allele (left panel). In contrast, the WT allele is hypermethylated and functional FBN1 mRNA is not sufficiently transcribed in the tissue or cell population (right panel). Open and closed circles represent unmethylated and methylated CpGs within the FBN1 CpG island shore, respectively; while black and white circles indicate either methylated or unmethylated CpGs. White and gray squares represent WT and KO alleles of FBN1, respectively.
Our previous study showed that the upstream region of the porcine FBN1, especially the CpG island shore, is highly homologous to the human FBN1, including the number of CpGs17. In the present study, we demonstrated variable DNA methylation levels within the FBN1 CpG island shore in several human cell lines. In addition, a significant correlation between DNA methylation and FBN1 expression was observed among human iPS cell lines. This suggests that human FBN1 is also likely to be regulated by DNA methylation within the FBN1 CpG island shore. Our findings on porcine FBN1 CpG island shore suggest a causal relationship between the expression level of FBN1 mRNA and the RM allele ratio of the WT allele in pigs. Therefore, this model can be applied to the characterization of pathogenic molecular mechanisms of Marfan syndrome in humans.
One cause of Marfan syndrome is haploinsufficiency, and Marfan patients with a low level of the FBN1 expression from a single WT allele have an increased risk of presenting several Marfan symptoms5. In addition, disease onset and severity differ among affected individuals with similar genetic abnormalities. Taken together with the results of this study, disease onset and severity of haploinsufficient Marfan syndrome involves DNA hypermethylation on the WT allele within the FBN1 CpG island shore that results in decreased functional FBN1 mRNA levels. Throughout development, the DNA methylation profile changes along with patterns of gene expression in developmental- and cell type-specific manners12,13. Abnormalities in epigenetics, including DNA methylation, are known to be involved in the onset of various diseases, such as cancer24. Thus, when accompanied by genetic mutation, stochastic epigenetic errors in haploinsufficient genes themselves during development and after birth are important factors that affect disease onset in heterozygous individuals.
Low levels of the FBN1 mRNA were observed in vivo in ear cartilage tissue from het KO pigs and fibroblast cells. In addition, FBN1 mRNA level was strongly correlated with RM allele ratios of the WT allele in ear cartilage samples, in consistent with in vitro fibroblast data. The SNP-containing het KO pigs we analyzed in the present study did not exhibit typical symptoms of Marfan syndrome during the experiment, and the decrease in mRNA levels were relatively mild. Furthermore, although differences in the expression levels of FBN1-related genes, such as TGFb and MMP2, were observed in our FBN1 het KO samples, these discrepancies were not identical to those observed in Marfan syndrome. This was likely due to the lack of overt phenotypes in the FBN1 het KO pigs analyzed. Autosomal recessive diseases can be simply reproduced in animal models by producing homozygotes of null alleles, whereas a difficulty in studying haploinsufficient autosomal dominant diseases is their variable penetrance and expressivity as is also the case for Marfan syndrome and its animal models of our FBN1 het KO pigs. Thus, one of the next issues to be addressed is whether SNP-containing FBN1 het KO pigs with apparent Marfan symptoms have both decreased FBN1 mRNA levels and RM allele ratios. The present study indicates that DNA hypermethylation of the FBN1 CpG island shore led to decreased mRNA levels, suggesting that artificial induction of DNA hypermethylation accelerates disease onset and manifestation of symptoms. Although epigenetics-modifying chemicals have mainly been studied for gene activation by targeting tumor suppressor genes for cancer therapy, the methyl-group donor, S-adenosyl methionine (SAM), or folate are possible candidates for the induction of DNA hypermethylation25. Thus, an alternative approach to investigating induced DNA hypermethylation of the FBN1 CpG island shore, which likely leads to efficient production of haploinsufficient disease models, is to feed FBN1 het KO pigs a high folate- or SAM-containing diet and see whether disease phenotypes manifest.
Our previous study on the epigenetic regulation of porcine preimplantation stage embryos suggested that DNA methylation of FBN1 is erased until the blastocyst stage and re-methylation occurs later during embryonic development17. In the present study, we found that the DNA methylation level of the FBN1 CpG island shore changed during the cell culture passages. These results suggest that DNA methylation status of the FBN1 CpG island shore dynamically changes depending on the developmental stage and cellular environment, and formation of DNA methylation patterns is crucial for the FBN1 expression in cells/tissues. In addition, the CpG island shore is located at the boundary of a constitutively unmethylated CpG island and constitutively methylated outside region, and this locational feature of the CpG island shore confers a variable outcome of random hypo- or hyper-methylation in addition to tissue/cell-type specific regulation. In the porcine and human FBN1 genes, the CpG island shore is located in close proximity to the transcriptional start site in the promoter region that is regulated by DNA methylation. Thus, FBN1-haploinsufficient Marfan syndrome including our Marfan model pigs may present early onset when the methylation pattern of the FBN1 CpG island shore of the WT allele is accidentally disturbed after birth to a DNA hypermethylation direction, resulting in decreased functional mRNA/protein levels in connective tissues. On the other hand, if such disturbances in DNA methylation patterns in the FBN1 CpG island shore occurs at very low levels in each cell or in very few cells in tissues, it is possible that Marfan syndrome disease onset could be delayed or not occur at all. Moreover regarding FBN1-dominant-negative Marfan syndrome, our finding that allele-dependent DNA methylation correlated with mRNA expression suggests that DNA hypomethylation of the mutant dominant-negative allele could exaggerate Marfan symptoms. Although any factors causing this type of ambiguity in FBN1 CpG island shore DNA methylation patterns are still not determined, ambiguous epigenetic errors within the FBN1 CpG island shore are a new insight into elucidating haploinsufficient Marfan syndrome onset with better accuracy.
In conclusion, DNA methylation of the FBN1 CpG island shore on the WT allele regulates functional FBN1 mRNA levels in FBN1 het KO cells/tissues. At present, hundreds of haploinsufficient diseases are known, but the mechanism(s) underlying regulation of the responsible genes leading to disease onset remain to be elucidated. Null mutations, in general, result in autosomal recessive genetic disease; however, the same type of null mutations in certain genes cause haploinsufficient dominant diseases, as is the case for Marfan syndrome. Induced DNA hypermethylation or epigenetic fluctuation of WT alleles as a novel modifier is likely a molecular contributor to haploinsufficiency that can further result in a wide variety of penetrance and expressivity of symptoms. About half of mammalian genes contain CpG islands in their promoter regions, and the expression of these genes can be regulated by stochastic changes of DNA methylation patterns within these CpG island shores. Therefore, it is important to characterize epigenetic fluctuations in the WT allele, together with conventional genetic null mutations in the KO allele, as part of the molecular pathogenesis of haploinsufficiency.
Materials and Methods
Animal care
All animal experiments were approved by the Institutional Animal Care and Use Committee of Meiji University (IACUC12-0008 and IACUC15-0001). All animal care and experimental procedures were performed in accordance with the Japan Act on Welfare and Management of Animals and all applicable regulations. The pigs were housed in a temperature-controlled room, had free access to water, and were provided with commercial feed appropriate for their growth stage (Nosan Co., Yokohama, Japan). For the autopsies, the pigs received an intramuscular injection of 1% mafoprazine mesylate (0.5 mg/kg, DS Pharma Animal Health Co., Ltd., Osaka, Japan) and midazolam (0.1 mg/kg, Takeda Pharmaceutical Co., Ltd., Tokyo, Japan), followed by an intravenous injection of sodium thiopental (Nipro ES Pharma Co., Ltd., Osaka, Japan). Anesthesia was maintained via the inhalation of isoflurane (DS Pharma Animal Health Co., Ltd.) while the pigs were exsanguinated.
Identification of a single nucleotide polymorphism (SNP) within the porcine FBN1 CpG island shore
In order to perform DNA methylation analysis of the FBN1 upstream region to distinguish the normal (WT) and FBN1-mutated null (KO) alleles, we attempted to find single nucleotide polymorphisms (SNPs) within the FBN1 CpG island shore by sequencing analysis. Briefly, genomic DNA was extracted from porcine peripheral blood using the NucleoSpin Blood kit (TaKaRa, Kyoto, Japan) according to the manufacture’s protocol. Purified genomic DNA was digested with HindIII (TaKaRa) and gel purified with a QIAquick Gel Extraction Kit (Qiagen, Hilden, Germany). Digested DNA was amplified with AccuPrime Taq DNA polymerase (Thermo Fisher Scientific, Carlsbad, CA) using specific primers for the porcine FBN1 CpG island shore region (FBN1_4F: 5′-GAGTTGAGGCAATGGGAAGA-3′, FBN1_4R: 5′-TGTGTCTGGGAGGCACAGT-3′). PCR was performed under the following conditions: 94 °C for 2 min; 35 cycles of 94 °C for 30 s, 60 °C for 30 s, and 68 °C for 1 min. and an adenine-tailing using Taq DNA polymerase at 72 °C for 10 min. Genomic PCR products were subcloned into the pGEM-T Easy vector (Promega, Madison, WI, USA), and sequencing was performed by Applied Biosystems 3130xL (Thermo Fisher Scientific).
Porcine FBN1 heterozygous knockout (het KO) fibroblast cell culture
Establishment of the FBN1 het KO fibroblast cell line was performed using porcine neonatal tail cartilage. Primary culture of porcine fibroblasts was performed as previously described26. Tail cartilage tissue was chopped into small pieces and placed in a type I collagen-coated dish containing minimum essential medium eagle (MEM) alpha modifications (Thermo Fisher Scientific) supplemented with 15% fetal bovine serum (HyClone, Thermo Fisher Scientific) and Penicillin-Streptomycin (Thermo Fisher Scientific). Fibroblast cells were grown in a humidified atmosphere with 5% CO2 at 37 °C, and sub-confluent cells were passaged and collected at the 6th and 20th passages (P6 and P20). Collected cells were stored at −80 °C until genomic DNA or RNA extraction.
Genomic DNA extraction and sodium bisulfite reaction
Genomic DNA extraction and bisulfite conversion were carried out as described previously14. Genomic DNA was purified from porcine fibroblast cells and ear cartilage using the DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer’s protocol. The genomic DNA was digested with HindIII (TaKaRa) and gel purified with a QIAquick Gel Extraction Kit (Qiagen), and bisulfite reactions were performed with the EZ DNA Methylation-Gold Kit (Zymo Research, Irvine, CA). Bisulfite-treated DNA was amplified with BioTaq HS DNA polymerase (BIOLINE, London, UK) using specific primers for the porcine FBN1 CpG island shore (FBN1_Bis_F: 5′-AGTTTTAATGTGAGTTGGATAAAAGGA-3′, FBN1_Bis_R: 5′-AAAAACTATACCACCTACACCAAAAA-3′). PCR was performed under the following conditions: 95 °C for 10 min; 40 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min; final extension 72 °C for 2 min. The PCR products were subcloned into pGEM-T Easy vector (Promega) for sodium bisulfite sequencing.
Gene expression analysis
Total RNA from porcine fibroblast cells and ear cartilage were extracted using the RNeasy Plus Mini and RNeasy Fibrous Tissue kits (Qiagen), respectively. First-strand cDNA synthesis was performed using a Superscript III First-strand Synthesis System (Thermo Fisher Scientific) with transcript-specific primers: FBN1_del1 (for FBN1 mRNA): 5′-GCACTCTCCACGGAGGTCC-3′, FBN1-del3 (for FBN1 preRNA): 5′-GCAATAGGCAAATTGAGAAACTTC-3′, and GAPDH: 5′-TCTGGGATGGAAACTGGAAG-3′. RT-PCR was performed using TaKaRa LA Taq (TaKaRa) with specific primers for FBN1 mRNA (FBN1_RT_F1: 5′-ATGTCCCTATGGTAGTTCCTGAGA-3′, FBN1_RT_R1: 5′-AGTTGGAATCCTTTGTTGCACT-3′), FBN1 preRNA (FBN1_RT_F3: 5′-CAGTCCCTCGACCACCAG-3′, FBN1_RT_R3: 5′-CCAACTTCATGACTTAAGCAGCAT-3′), and GAPDH (GAPDH_RT_F: 5′-ACCACAGTCCATGCCATCAC-3′, GAPDH_RT_R: 5′-TCCACCACCCTGTTGCTGTA-3′). RT-PCR was performed using the following conditions for fibroblast cells: 95 °C for 5 min; 26 cycles (FBN1 mRNA), 33 cycles (FBN1 preRNA), or 23 cycles (GAPDH) of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min; final extension 72 °C for 10 min. For ear cartilage samples, PCR were performed with 28 cycles (FBN1 mRNA), 33 cycles (FBN1 preRNA), or 26 cycles (GAPDH).
Gene expression array data were deposited in the Gene Expression Omnibus repository (GEO; http//www.ncbi.nlm.nih.gov/geo/) with the following accession numbers: GSE20750, GSE24677, and GSE4209923,27. All expression data were analyzed using an Agilent-014850 Whole Human Genome Microarray 4×44 K G4112F and extracted for the specific FBN1 gene probe (A_23_P65678). The expression data were processed by quantile normalization for analysis.
DNA methylation analysis of the FBN1 CpG island shore region in human somatic cells
DNA methylation data of five human somatic cell lines (MRC5, AM936EP, Edom22, PAE551, and UtE1104) and the corresponding iPS cell lines established from these somatic cells were collected from our previous study using an Infinium HumanMethylation450K BeadChip28,29. The data were extracted for specific probes that were located outside (probe I, cg01529092) or within (Probe II, cg14164380) the CpG island shore, as well as inside the CpG island (probe III, cg15385562). These methylation data were deposited in GEO with the following accession numbers: GSE73938, GSE99716, GSE116924, and GSE141521.
Statistical analysis
All experiments were performed in triplicate, and the results were represented as mean values with standard deviation. All statistical analyses were performed using GraphPad Prism 8.0 software (San Diego, CA, USA). Statistical significance of two independent experiments were determined using Student’s t-test, and p < 0.05 were defined as significant.
References
Judge, D. P. et al. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J. Clin. Invest. 114, 172–181 (2004).
Jondeau, G. et al. The translational science of Marfan syndrome. Heart 97, 1206–1214 (2011).
Robinson, P. N. et al. Mutations of FBN1 and genotype-phenotype correlations in Marfan syndrome and related fibrillinopathies. Hum. Mutat. 20, 153–161 (2002).
Potter, K. J. et al. The c.7409G > A (p.Cys2470Tyr) Variant of FBN1: Phenotypic Variability across Three Generations. Mol. Syndromol. 4, 125–135 (2013).
Aubart, M. et al. The clinical presentation of Marfan syndrome is modulated by expression of wild-type FBN1 allele. Hum. Mol. Genet. 24, 2764–2770 (2015).
Cook, D. L. et al. Modeling stochastic gene expression: implications for haploinsufficiency. Proc. Natl Acad. Sci. USA 95, 15641–15646 (1998).
Umeyama, K. et al. Generation of heterozygous fibrillin-1 mutant cloned pigs from genome-edited foetal fibroblasts. Sci. Rep. 6, 24413 (2016).
Shiota, K. DNA methylation profiles of CpG islands for cellular differentiation and development in mammals. Cytogenet. Genome Res. 105, 325–334 (2004).
Lieb, J. D. et al. Applying whole-genome studies of epigenetic regulation to study human disease. Cytogenet. Genome Res. 114, 1–15 (2006).
Ohgane, J. et al. Epigenetics: the DNA methylation profile of tissue-dependent and differentially methylated regions in cells. Placenta 29, S29–35 (2008).
Reik, W. et al. Epigenetic reprogramming in mammalian development. Science 293, 1089–1093 (2001).
Shiota, K. et al. Epigenetic marks by DNA methylation specific to stem, germ and somatic cells in mice. Genes. Cell 7, 961–969 (2002).
Sakamoto, H. et al. Cell type-specific methylation profiles occurring disproportionately in CpG-less regions that delineate developmental similarity. Genes Cells 12, 1123–1132 (2007).
Arai, Y. et al. DNA methylation profiles provide a viable index for porcine pluripotent stem cells. Genesis 51, 763–776 (2013).
Arai, Y. et al. Ultra-Deep Bisulfite Sequencing to Detect Specific DNA Methylation Patterns of Minor Cell Types in Heterogeneous Cell Populations: An Example of the Pituitary Tissue. PLoS One 11, e0146498 (2016).
Takeda, N. et al. Impact of pathogenic FBN1 variant types on the progression of aortic disease in patients with Marfan syndrome. Circ. Genom. Precis. Med. 11, e002058 (2018).
Arai, Y. et al. Establishment of DNA methylation patterns of the Fibrillin1 (FBN1) gene in porcine embryos and tissues. J. Reprod. Dev. 63, 157–165 (2017).
Ashikaga, T. et al. Altered expression of cyclic nucleotide phosphodiesterase isozymes during culture of aortic endothelial cells. Biochem. Pharmacol. 54, 1071–1079 (1997).
Danesh Mesgaran, S. et al. mRNA expression pattern of selected candidate genes differs in bovine oviductal epithelial cells in vitro compared with the in vivo state and during cell culture passages. Reprod. Biol. Endocrinol. 14, 44 (2016).
Reeves, S. R. et al. Stability of gene expression by primary bronchial epithelial cells over increasing passage number. BMC Pulm. Med. 18, 91 (2018).
Sakai, L. Y. et al. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J. Cell Biol. 103, 2499–2509 (1986).
Pfaff, M. et al. Cell adhesion and integrin binding to recombinant human fibrillin-1. FEBS Lett. 384, 247–250 (1996).
Nishino, K. et al. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet. 7, e1002085 (2011).
Ushijima, T. & Okochi-Takada, E. Aberrant methylations in cancer cells: where do they come from? Cancer Sci. 96, 206–211 (2005).
ElGendy, K. et al. Effects of dietary interventions on DNA methylation in adult humans: systematic review and meta-analysis. Br. J. Nutr. 120, 961–976 (2018).
Kurome, M. et al. Production efficiency and telomere length of the cloned pigs following serial somatic cell nuclear transfer. J. Reprod. Dev. 54, 254–258 (2008).
Tateno, H. et al. Podocalyxin is a glycoprotein ligand of the human pluripotent stem cell-specific probe rBC2LCN. Stem Cell Transl. Med. 2, 265–273 (2013).
Nishino, K. et al. Epigenetic-scale comparison of human iPSCs generated by retrovirus, Sendai virus or episomal vectors. Regen. Ther. 9, 71–78 (2018).
Takasawa, K. et al. DNA hypermethylation enhanced telomerase reverse transcriptase expression in human-induced pluripotent stem cells. Hum. Cell 31, 78–86 (2018).
Acknowledgements
Financial support for this study was provided by Grant-in-Aid for Scientific Research (A) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to HN and JO), Research Project Grant (B) by Institute of Science and Technology Meiji University (to JO), and Grant-in-Aid for Scientific Research (C) (to YA). This work is supported in part by a Research Funding for Computational Software Supporting Program from Meiji University, Meiji University International Institute for Bio-Resource Research, and Leading Advanced Project for medical innovation (LEAP), AMED. We thank Dr Michael Rehli for kindly providing us with the pCpG-L vector for luciferase assay. YA thanks Dr Ken Takasawa for his kind cooperation with this project.
Author information
Authors and Affiliations
Contributions
Y.A. and J.O. conceived and designed experiments and research; Y.A. and J.O. wrote the paper; Y.A., K.U., N.O., K.N. and N.K. performed experiments; Y.A., K.N. and J.O. analyzed data; Y.A., K.U., H.N. and J.O. discussed the results. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Arai, Y., Umeyama, K., Okazaki, N. et al. DNA methylation ambiguity in the Fibrillin-1 (FBN1) CpG island shore possibly involved in Marfan syndrome. Sci Rep 10, 5287 (2020). https://doi.org/10.1038/s41598-020-62127-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-62127-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
