
Abstract
Imprinted genes are expressed from only one allele in a parent of origin–specific manner. The cyclin-dependent kinase inhibitor p57kip2 is encoded by an imprinted gene Cdkn1c, with the paternal allele being silenced. The possible expression and function of the paternal allele of Cdkn1c have remained little studied, however. We now show that the paternal allele of the Cdkn1c gene is expressed at a low level in the developing mouse neocortex. Surprisingly, the central nervous system-specific conditional deletion of the paternal allele (pat cKO) at the Cdkn1c locus resulted in a marked reduction in brain size. Furthermore, pat cKO gradually reduced the number of neural stem-progenitor cells (NPCs) during neocortical development, and thus reduced the number of upper-layer neurons, which were derived from late-stage NPCs. Our results thus show that the paternal allele of the Cdkn1c locus plays a key role in maintenance of NPCs during neocortical development.
Similar content being viewed by others


Introduction
Genomic imprinting refers to an epigenetic process that results in the inactivation of one of the two alleles of a gene in a parent of origin–dependent manner1,2,3. It is achieved mainly by allele-specific DNA methylation at a subset of CpG islands, known as imprinting control regions (ICRs), during early developmental stages4. Imprinted genes play essential roles in development, homeostasis, and behavior in mammals5,6. Changes at imprinted gene loci in humans are associated with diseases such as Beckwith-Wiedemann syndrome, Prader-Willi syndrome, and Angelman syndrome7,8, many of which are characterized by altered growth and mental disorders6. Although canonical genomic imprinting has been thought to result in the complete silencing of one allele of a gene, recent studies have shown that silencing of some imprinted genes appears to be incomplete or reversed to various extents in the brain9. For instance, derepressed expression of the imprinted alleles of Igf2 and Dlk1 contributes to the regulation of adult neural stem cells in mice10,11. However, the functions of the imprinted alleles of other genes remain mostly obscure.
The Cdkn1c gene is imprinted, with the maternal allele being expressed, and is located in the distal region of mouse chromosome 7 and human chromosome 11p1512,13. In mice, DNA methylation of two ICRs, KvDMR14,15,16 and ICG517, has been suggested to suppress expression of the paternal allele of the Cdkn1c. The Cdkn1c-encoded protein p57kip2 is a cyclin-dependent kinase inhibitor (CKI)18,19 that is highly expressed in neural and skeletomuscular tissues20,21 during embryonic development. In humans, changes at the Cdkn1c gene locus are associated with Beckwith-Wiedemann syndrome, features of which include excessive growth and an increased risk of childhood cancer22,23. Gene knockout (KO) studies have implicated p57kip2 in regulation of fetal growth and placental development20,24,25. In the central nervous system (CNS), p57kip2 plays a key role in regulation of the proliferation and differentiation of embryonic neural stem-progenitor cells (NPCs) and adult neural stem cells26,27,28,29,30,31,32,33. CNS-specific conditional KO of the maternal Cdkn1c allele resulted in the induction of cell death in a manner dependent on the transcriptional regulators E2F1 and p53, thinning of the neocortex, and pronounced hydrocephalus in mice, with the latter effects possibly reflecting a function of p57kip2 in the subcommissural organ (SCO)30.
The paternal allele of the Cdkn1c gene has been thought to be completely silenced, given that conventional KO of the maternal allele appeared to result in elimination of Cdkn1c mRNA and protein20 and that a reporter for the paternal allele did not show any expression during normal development unless challenged by stress34. Indeed, the phenotype of conventional maternal Cdkn1c KO mice appears essentially identical to that of conventional null Cdkn1c KO mice20,24,32, and paternal Cdkn1c KO mice have been found to manifest no distinct phenotype24. These observations have thus suggested that only the maternal allele of the Cdkn1c gene locus is indispensable. However, it has remained possible that the paternal allele of this locus is transcribed at a low level, and the necessity of the paternal allele for development has remained poorly explored.
Here we examined the expression and functional importance of the paternal Cdkn1c allele in the developing mouse brain. Reverse transcription (RT) and allele-specific quantitative polymerase chain reaction (qPCR) analysis revealed that the paternal allele is indeed expressed at a low level in embryonic NPCs. Unexpectedly, CNS-specific paternal Cdkn1c KO resulted in a substantial reduction in brain size, with the number of upper layer neurons in the neocortex showing a marked decrease. Consistent with this latter observation, the number of NPCs that give rise to upper layer neurons was also decreased in the mutant mice. Our findings thus uncover an essential role for the imprinted allele of the Cdkn1c gene in the developing nervous system.
Results
Expression of the paternal Cdkn1c allele in neocortical NPCs and neurons during mouse embryonic development
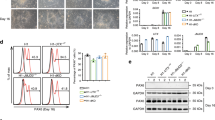
We first examined whether expression of the paternal (imprinted) allele of the Cdkn1c gene could be detected in the developing brain with the use of chimeric mice derived from a cross between lines—C57BL/6J (BL6) and JF1/Ms (JF1)—that differ with regard to single nucleotide polymorphisms (SNPs) at this locus. We thus collected F1 hybrid embryos at embryonic day (E) 16 from a cross between JF1 male and BL6 female mice. Total RNA isolated from the embryonic neocortex was then subjected to RT and allele-specific qPCR analysis with primers designed to recognize individual SNPs (Fig. 1a). In contrast to previous results showing that the paternal allele of the Cdkn1c gene is completely silenced20,34, we detected expression of the paternal allele at a level corresponding to ~1% to 2% of that of the maternal allele (Fig. 1b). We did not detect Cdkn1c mRNA in embryos from a cross between BL6 male and BL6 female mice with the use of JF1-specific primers or in those from a cross between JF1 male and JF1 female mice with the use of BL6-specific primers, confirming the specificity of the line-specific primers. We also performed the reciprocal cross (BL6 male with JF1 female) and examined offspring for expression of the maternal and paternal Cdkn1c alleles by RT-qPCR analysis. The expression level of the paternal allele relative to that of the maternal allele was almost the same as that for offspring of JF1 male with BL6 female (Supplementary Fig. 1a), indicating that this paternal expression was not due to a strain specific effect. Furthermore, the paternal and maternal Cdkn1c expression levels reported in the previous RNA sequencing analysis in the cortex at E13 and P0 are consistent with our results35. We next deleted the paternal allele of the Cdkn1c gene in the CNS by crossing JF1 female mice heterozygous for a Cre recombinase transgene under the control of the Nestin enhancer (Nestin-Cre) with BL6 male mice harboring floxed alleles of the Cdkn1c gene (Cdkn1fl/fl) (Fig. 1c and Supplementary Fig. 1d). Expression of the paternal Cdkn1c allele in the neocortex was partially but significantly reduced in the resulting conditional KO (cKO) embryos at E16 compared with control littermates lacking the Nestin-Cre transgene (Fig. 1c). Deletion of the maternal Cdkn1c allele by the Nestin-Cre reduced the expression of maternal Cdkn1c to a similar level with that observed in the paternal cKO (Supplementary Fig. 1b,c). The partial reduction of paternal and maternal Cdkn1c transcripts by these cKO is likely due to those expressed before the expression of Nestin-Cre, which starts to be expressed in the CNS from ~E1136. This result thus suggests that the paternal Cdkn1c allele indeed contributed to the Cdkn1c transcripts detected with BL6-specific primers in these embryos. We then examined expression of the paternal Cdkn1c allele in NPCs and neurons isolated from the neocortex of E16 embryos derived from a cross between BL6 male and JF1 female mice. These cells were isolated as CD133+CD24− and CD133−CD24+ fractions, respectively, by fluorescence-activated cell sorting (FACS), and both were found to express the paternal Cdkn1c allele (Fig. 1b). Together, these results thus indicated that the paternal allele of the Cdkn1c gene is transcribed, albeit at a low level, in the developing neocortex.

Detection of Cdkn1c mRNA expression from the paternal allele in the embryonic neocortex. (a) Scheme of allele specific qPCR of Cdkn1c. F1 hybrid embryos of C57BL/6J (BL6) female and JF1/MS (JF1) male parents have SNPs derived from their parents. qPCR was performed by the use of specific primers for each allele. (b) The ratio of paternal to maternal Cdkn1c mRNA expression in NPCs and in neurons was determined by allele specific qPCR under the hybrid (JF1 male and BL6 female) genetic background at E16. FACS was used to isolate NPCs (CD133+CD24− population) and neurons (CD133−CD24+ population). (c,d) Allele specific qPCR of Cdkn1c mRNA in the neocortex isolated from control (Cdkn1c pat flox (BL6)/+(JF1)) and hybrid paternal Cdkn1c cKO (Nestin-Cre; Cdkn1c pat flox(BL6)/+(JF1)) mice at E16. Cdkn1c mRNA expression from each allele was normalized to β-actin. Data are expressed relative to the corresponding value for control mice. (e) qPCR of total Cdkn1c mRNA expression in the neocortex isolated from control and paternal Cdkn1c cKO mice in the BL/6J background at P0. Cdkn1c mRNA expression was normalized to β-actin. Data are expressed relative to the corresponding value for control mice. Data are mean + s.e.m from three independent experiments. Paired two-tailed Student’s t-test; **P < 0.01. n.s., not significant.
We investigated whether deletion of the paternal Cdkn1c allele might affect expression of the maternal allele. However, the amounts of maternal or total Cdkn1c mRNA in the neocortex did not differ significantly among E16 embryos derived from a cross between BL6 male Cdkn1cfl/fl and JF1 female Nestin-Cre+/− mice (Fig. 1d,e). We also did not detect any significant differences in the total protein level of p57kip2 after the paternal Cdkn1c deletion (Supplementary Fig. 2d).
We next examined the directionality of transcription of Cdkn1c by performing Northern blot analysis of control brains. As shown in the Supplementary Fig. 2a,b, we detected transcripts only generated from the sense (coding) direction, but not those generated from the antisense direction. We next performed direction (strand)–sensitive RNA-sequencing analysis to further examine the directionality of Cdkn1c transcripts. This analysis again showed that almost all of the reads (1766 out of 1778) that mapped to the Cdkn1c locus were in the sense (coding) direction (Supplementary Fig. 3). At the exon 4 locus at which we conducted allele specific qPCR in Fig. 1, no read was mapped to the antisense strand (0 out of 650). This high ratio of transcripts from the sense direction at the exon 4 suggests that transcripts derived from this locus of the paternal allele are also synthesized from the sense (coding) strand similar to the maternal one, although this does not exclude the possibility of antisense expression at the paternal Cdkn1c allele. We also compared the pattern of Cdkn1c transcripts between control and paternal Cdkn1c cKO brain. The pattern of reads that mapped to the Cdkn1c locus was largely unchanged by paternal Cdkn1c cKO (Supplementary Fig. 3), again suggestive of the absence (or low abundance) of paternal-specific nonconventional Cdkn1c transcripts.
We then examined the possibility that the paternal Cdkn1c deletion may affect other imprinted genes located close to Cdkn1c in the same gene cluster. We performed RT-qPCR analysis for such genes (Cd81, Phlda2, Osbp15 and Nap1l4) and found that the expression of these genes was not largely affected by the paternal Cdkn1c deletion (Supplementary Fig. 4a–d). We also performed bisulfite sequencing of KvDMR1 and ICG5, located near Cdkn1c, in the control and paternal Cdkn1c cKO neocortices at E16. The levels of DNA methylation at these loci were slightly increased but not overtly affected by paternal Cdkn1c cKO (Supplementary Fig. 4e,f). We thus concluded that the levels of imprinting in the same gene cluster were not significantly affected by the paternal Cdkn1c cKO.
CNS-specific paternal Cdkn1c KO reduces the brain size and the number of upper layer neurons at postnatal stage
We next investigated whether the paternal Cdkn1c allele plays a role in mouse brain development. We deleted the paternal allele in a CNS-specific manner by crossing BL6 Cdkn1cfl/fl male with BL6 Nestin-Cre+/− female mice and found that such deletion resulted in a substantial reduction in brain size apparent at postnatal day (P) 60 (Fig. 2a). This finding was unexpected given the low level of expression of the paternal allele compared with the maternal allele. Immunohistofluorescence analysis of coronal sections revealed that this size reduction was apparent throughout the entire forebrain—including the neocortex, basal ganglia, thalamus, and septal nuclei—of the paternal cKO mice at P24 (Fig. 2b). The total section size of the paternal Cdkn1c cKO brain at the level of 0.38 to 1.18 mm relative to the bregma was only 72.37 ± 6.07% (mean ± s.e.m., n = 3 mice) of that for the control brain at P24. With regard to the neocortex, the mediolateral surface length at this level relative to the bregma was reduced to 81.44 ± 1.53% (mean ± s.e.m., n = 3 mice) for the paternal Cdkn1c cKO brain compared with the control brain at P24. The thickness of the primary somatosensory area (S1) of the paternal Cdkn1c cKO brain was also reduced to 71.33 ± 10.00% (mean ± s.e.m., n = 3 mice) of that for the control brain at P24.

CNS-specific deletion of the Cdkn1c paternal allele reduced the brain size and the number of upper layer neurons at P24. (a–g) Cdkn1c paternal cKO (Nestin-Cre; Cdkn1c pat flox/+) and control (Cdkn1c pat flox/+) mice were sacrificed at P60 (a) and P24 (b–g). (a) Dorsal view of Cdkn1c paternal cKO and control brains. (b) Immunofluorescence staining of Cux1 (green) and Ctip2 (red) in coronal sections of Cdkn1c paternal cKO and control mice. Nuclei were stained with Hoechst (blue). (c) Higher magnifications of the neocortical somatosensory area in (b) (left panel). Quantitative analysis of cells positive for Cux1 and Ctip2 per area within 200 μm wide bins (right panel) (n = 3 mice for each genotype). (d–g) The number of total cells (Hoechst+) (d), neurons (NeuN+) (e), non-neuronal cells (NeuN−) (f) and glial cells (S100β+) (g) per 200 μm wide bins in the neocortical area were quantified (n = 4 mice for each genotype). Data are mean + s.e.m. Unpaired two-tailed Student’s t-test; **P < 0.01 n.s., not significant. Scale bars: 500 μm in (a); 250 μm in (b); 100 μm in (c).
The forebrain phenotype of the paternal Cdkn1c cKO mice appeared largely similar to that of CNS-specific maternal Cdkn1c cKO mice previously generated30, although the former was generally less pronounced and showed some distinct features. One such feature of the paternal cKO brain was the apparent absence of hydrocephalus, one of the most remarkable characteristics of the maternal Cdkn1c cKO brain30. The ventricles of the paternal Cdkn1c cKO brain were not as expanded as those of the control brain at P0 or P24 (Figs. 2b and 3a). The previous study of maternal Cdkn1c cKO mice concluded that the thinning of the neocortex also apparent in these mice was a result of the hydrocephalus caused by impaired development of the SCO30. Our results revealing a thin neocortex without apparent hydrocephalus in the paternal Cdkn1c cKO brain thus indicates that this neocortical phenotype is not due to hydrocephalus, at least not in these animals.

CNS-specific deletion of the Cdkn1c paternal allele reduced the number of upper layer neurons at P0 and progenitors at E16. (a) Immunofluorescence staining of Cux1 (green) and Ctip2 (red) in coronal sections of Cdkn1c paternal cKO and control mice at P0. Nuclei were stained with Hoechst (blue). (b) Higher magnifications of neocortical somatosensory area in (a) (left panel). Quantitative analysis of cells positive for Cux1 and Ctip2 per area within 200 μm wide bins (right panel) (n = 4 pups for each genotype). (c–f) Immunofluorescence staining of Pax6 (c,e) and Tbr2 (d,f) at E16 (c,d) and E13 (e,f), respectively. The cells positive for Pax6 and Tbr2 per area within 200 μm wide bins in the neocortical region were quantified (n = 4 embryos for each genotype in c,d and n = 3 embryos for each genotype in e,f). Data are mean + s.e.m. Unpaired two-tailed Student’s t-test; *P < 0.05. n.s., not significant. Scale bars: 500 μm in (a); 100 μm in (b); 50 μm in (c–f).
We next examined the neocortex of paternal Cdkn1c cKO mice at P24 in more detail. Consistent with the reduced thickness of the neocortex, the cell number in S1 was reduced in the paternal Cdkn1c cKO mice compared with control mice (Fig. 2d). The number of NeuN+ neurons was reduced by paternal Cdkn1c cKO (Fig. 2e) while the number of NeuN− cells and S100β+ astrocytes was not (Fig. 2f,g). Among neurons, the number of Cux1+ upper layer neurons was significantly reduced whereas that of Ctip2+ deep layer neurons was not (Fig. 2c). This selective reduction in the number of upper layer neurons relative to deep layer neurons in the paternal Cdkn1c cKO neocortex at P24 was confirmed by the detection of a reduced thickness of the upper layers but not of the deep layers in S1 (Supplementary Fig. 5a). We observed similar but severer phenotypes in the maternal Cdkn1c cKO mice at P14 except the reduction of NeuN− cells (Supplementary Fig. 6). Our results together suggested that the paternal Cdkn1c allele (as well as its maternal allele) might contribute to the genesis of specific neuronal subtypes.
Paternal Cdkn1c cKO reduces the number of neocortical NPCs at late embryonic stages
We next examined when and how paternal Cdkn1c cKO results in a reduction in the number of upper layer neurons in the neocortex during development. We first investigated the neocortex of control and paternal Cdkn1c cKO mice at P0 and found that the reduction in the number of Cux1+ upper layer neurons was already apparent at this stage (Fig. 3a,b). Of note, the number of Ctip2+ deep layer neurons at this stage was actually increased by paternal Cdkn1c cKO (Fig. 3b). Consistent with these results, the thickness of the upper layers was reduced but that of the deep layers tended to increase in the paternal Cdkn1c cKO mice at P0 (Supplementary Fig. 5b).
To investigate the mechanism underlying the selective reduction in the number of upper layer neurons in the postnatal neocortex of paternal Cdkn1c cKO mice, we evaluated the production of deep layer and upper layer neurons at embryonic stages. We first examined the numbers of Pax6+ NPCs and Tbr2+ intermediate neuronal progenitors (INPs) in S1 at E13 and found no significant difference between control and paternal Cdkn1c cKO mice (Fig. 3e,f). However, paternal Cdkn1c cKO resulted in a significant reduction in the numbers of both these cell types at E16 (Fig. 3c,d). Consistent with these findings, the number of proliferating cells positive for Ki67 was also reduced in the neocortex of paternal Cdkn1c cKO mice at E16 but not at E13 (Fig. 4a,b). Given that most deep layer neurons and upper layer neurons in S1 are born before and after E13.5, respectively, the differences in the numbers of NPCs and INPs apparent at E16 but not at E13 may explain, at least in part, the selective reduction in the number of upper layer neurons induced by paternal Cdkn1c cKO. We also found that the number of apoptotic cells positive for the cleaved form of caspase-3 was increased among NPCs in the neocortex of paternal Cdkn1c cKO mice at both E13 and E16 (Fig. 4c,d). Related to apoptosis induction, we also detected increased expression of the gene for the CKI Cdkn1a (p21Cip1), a target of p53, in the neocortex of paternal Cdkn1c cKO mice at E16 (Fig. 4e). The increase in the extent of cell death might thus account for the reduction in the number of NPCs and subsequently that in INPs in the paternal Cdkn1c cKO neocortex. We then performed transcriptomic analysis of the control and paternal Cdkn1c cKO neocortices by RNA sequencing and gene ontology (GO) analysis on differentially expressed genes. Consistent with the increased cell death, we observed enrichment of GO terms such as neuronal death, DNA damage response and signal transduction by p53 class mediator in the upregulated genes by paternal Cdkn1c deletion (Supplementary Fig. 7a). Also, according to the RNA sequencing results, the apoptotic genes such as Ccng1, Trp73 and Sesn2 were significantly upregulated in paternal Cdkn1c cKO neocortex compared to control. On the other hand, the GO terms enriched in the downregulated genes by paternal Cdkn1c deletion include those related to the mitochondrion pathway such as oxidative phosphorylation (Supplementary Fig. 7b). Together, these results indicate that the paternal Cdkn1c allele promotes cell survival and plays an important role in the generation of appropriate numbers of NPCs, INPs, and cortical neurons during development.

CNS-specific deletion of the Cdkn1c paternal allele reduced the number of proliferating progenitors at E16 and increased the number of apoptotic cells at E13 and E16. (a,b) Immunofluorescence staining of Ki67 at E13 (a) and E16 (b) (left panel). Quantitative analysis of cells positive for Ki67 per area within 200 μm wide bins (right panel) (n = 3–4 embryos for each genotype). Data are mean + s.e.m. Unpaired two-tailed Student’s t-test. (c) Immunostaining of cleaved caspase-3 at E13. The boxed regions in the left panels are shown at a higher magnification in the right panel. Yellow arrowheads indicate cleaved caspase-3 positive cells. (d) The number of cleaved caspase-3 positive cells per area within 500 μm wide bins were quantified at E13 and E16 (n = 4–6 embryos for each genotype). Data are mean + s.e.m. Unpaired two-tailed Student’s t-test. (e) qPCR of Cdkn1a mRNA expression in the neocortex isolated from control and paternal Cdkn1c cKO mice at P0. Cdkn1a mRNA expression was normalized to β-actin (n = 3 independent experiments). Data are mean + s.e.m.; expressed relative to the corresponding value for control mice. Paired two-tailed Student’s t-test. *P < 0.05. n.s., not significant. Scale bars: 50 μm in (a,b); 100 μm in (c).
Discussion
We have here detected a low level of expression of the paternal Cdkn1c allele in the developing mouse neocortex, in contrast to previous findings that the paternal allele is completely imprinted and silenced20,34. Immunohistochemical analysis thus previously showed that the amount of p57kip2 (Cdkn1c) protein was reduced to an undetectable level in the brain by deletion of the maternal Cdkn1c allele34, and reporter mice that allow monitoring of the expression of the paternal allele on the basis of the activity of firefly luciferase encoded by a construct knocked-in at this locus did not show a luminescence signal under normal dietary conditions34. However, in contrast to the allele-specific qPCR analysis adopted in our study, expression of the paternal Cdkn1c allele might have been too low compared with that of the maternal allele to have been detected by these previous methods.
Although expression of the paternal Cdkn1c allele in the brain was <2% of that of the maternal allele at E16, we found that paternal Cdkn1c cKO resulted in prominent changes to the brain including thinning of the neocortex. Such thinning previously observed in maternal Cdkn1c cKO mice was suggested to be a result of defects in the SCO30. However, the neocortical thinning detected in our paternal Cdkn1c cKO mice appears to be independent of any effect on the SCO, given the absence of hydrocephalus. Instead, our results suggest that increased cell death and a consequent reduction in the number of neocortical NPCs at the late stage of development give rise to a reduced level of neurogenesis and thinning of the neocortex in paternal Cdkn1c cKO mice. Our results have thus revealed an essential role for the paternal Cdkn1c allele in control of the survival of neocortical NPCs and the genesis of neurons—in particular, upper layer neurons produced at the late stage of development.
How the paternal Cdkn1c allele fulfills this role despite its low expression level remains unknown. It is possible that the paternal allele is expressed at similar or even higher levels relative to the maternal allele in a subpopulation of NPCs in which it plays a protective role. Alternatively, the paternal and maternal alleles might confer expression of different mRNA or protein isoforms, although we did not detect any difference in total Cdkn1c mRNA and protein levels in the absence or presence of the paternal allele. The paternal allele thus does not appear to affect the expression level of the maternal allele. It is also possible that transcripts other than Cdkn1c mRNA are produced from the paternal Cdkn1c allele or from cis- or trans-regulatory elements within the paternal Cdkn1c locus and contribute to the changes to the brain apparent in paternal Cdkn1c cKO mice.
Prenatal protein restriction and intrauterine growth retardation have been associated with various psychiatric conditions including schizophrenia, attention-deficit and hyperactivity disorder, autism spectrum disorder, and major affective depression37. Of interest in this regard, restriction of maternal dietary protein during pregnancy induces demethylation and aberrant induction of the Cdkn1c locus in the prefrontal cortex and mesolimbic dopaminergic system of the resulting offspring38. Indeed, such protein restriction was found to result in permanent derepression of the imprinted paternal Cdkn1c allele through a folate-dependent mechanism of DNA methylation loss34. However, whether or how such derepression of the paternal Cdkn1c locus affects brain development remains to be clarified. Given that we found that the paternal Cdkn1c allele plays an essential role in generation of appropriate numbers of NPCs and neurons, derepression of this allele induced by a low-protein diet may have an impact on NPC proliferation and neurogenesis during neocortical development. We also found that paternal Cdkn1c cKO resulted in a reduction in the staining intensity for tyrosine hydroxylase (TH) in the striatum (data not shown). Given that p57kip2 (Cdkn1c) has been implicated in the production of TH+ dopaminergic neurons27 and that twofold overexpression of Cdkn1c conferred by a transgene both increased TH expression in the brain and altered behaviors that are dependent on the mesolimbic dopaminergic system39,40, derepression of the paternal Cdkn1c locus induced by a low-protein diet might thus lead to behavioral changes through modulation of neurogenesis in both the neocortex and mesolimbic dopaminergic system. The possible role of derepression of the paternal Cdkn1c allele in the link between early-life adversity and aberrant brain development associated with psychiatric disorders thus warrants further investigation.
Experimental Procedures
Mice
C57BL6/J (BL6) mice were purchased from CLEA Japan. JF1/Ms (JF1) mice were provided by RIKEN BRC (RBRC00639) through the National BioResource Project of the MEXT/AMED, Japan, and purchased from National Institute of Genetics, Japan. Nestin-Cre mice and Cdkn1c floxed mice were kindly provided by Ryoichiro Kageyama and Keiichi Nakayama, respectively. To generate JF1 Nestin-Cre mice, Nestin-Cre mice were backcrossed to JF1 mice, and confirmed the SNPs at Cdkn1c genomic locus with sanger sequencing. All mice were maintained and studied according to protocols approved by the Animal Care and Use Committee of The University of Tokyo (approval numbers: P25-8, P25-27, PH27-3 and P30-4).
FACS
Neocortex was prepared by manual dissection, digested enzymatically with a papain-based solution (Sumitomo Bakelite) and then resuspended in 0.3% BSA/PBS containing primary antibodies (PE-conjugated CD133 (mouse, 1:500, BioLegend, 141204) and APC-conjugated CD24 (mouse, 1:500, BioLegend, 101814)). Cells were then sorted with FACS Aria IIIu (BD). Debris and aggregated cells were gated out by forward and side scatter. Gating was done with isotype controls. NPCs and neurons were isolated as CD133+CD24− fraction and CD133−CD24+ fraction, respectively.
RNA extraction and RT-qPCR
Allele specific qPCR was performed on cDNA prepared from sorted cells or neocortical tissues of F1 hybrid mice obtained by crossing BL6 and JF1. For other primer sets, qPCR was carried out with the use of cDNA prepared from neocortical tissues of BL6 mice. Total RNA was extracted using RNAiso Plus (Takara) following the instructions of the manufacturer. Reverse transcription (RT) was performed with at the maximum of 0.5 μg of total RNA and ReverTra Ace qPCR Master Mix with gDNA remover (TOYOBO). The obtained cDNA was subjected to real-time PCR analysis in a Roche LightCycler 480 II with THUNDERBIRD SYBR qPCR kit (TOYOBO). As for allele specific qPCR, plasmid DNA containing each SNPs was used as a standard and relative copy number was calculated according to the molecular weight. In other experiments, the amount of mRNA quantified was normalized relative to that of β-actin mRNA.
The used primers were as follows:
Actb
Fw AATAGTCATTCCAAGTATCCATGAAA
Rv GCGACCATCCTCCTCTTAG
Cdkn1a
Fw CACCTCTAAGGCCAGCTA
Rv AGCAATGTCAAGAGTCGG
Cd81
Fw GGAAGCTGTACCTCATTGGA
Rv CAGCACCATGCTCAGAATC
Phlda2
Fw TCACCATCGTCACCAACTA
Rv CGGTTCTGGAAGTCGATCA
Osbp15
Fw ACCGGCTAAAGATGCTACAA
Rv TGGAGCTTAACATGGTCGAA
Nap1l4
Fw CGCACATAGAAGCTAAGTTCTAC
Rv TCCTCATTCTCACTGTGCC
Cdkn1c
Common GGGCAGTACAGGAACCATTTC
BL6 TTAGCTTACAGTGTCCCGCA
JF1 TTAGCTTACAGTGTCCCGTA
Allele specific qPCR primers were designed as previously described41. BL6/JF1 SNP sites (BL6 chr7: 143458462, NC_000073, GenBank) were identified using NIG Mouse Genome Database (http://molossinus.nig.ac.jp/msmdb/index.jsp). Total Cdkn1c mRNA was quantified with common and BL6 primers.
Immunohistochemistry
For immunofluorescence staining, brains were postfixed with ice-cold 4% (wt/vol) paraformaldehyde (PFA) at 4 °C for 2 h, equilibrated with 30% (w/v) sucrose in PBS, and frozen in OCT (Tissue TEK). Coronal sections (15–16 μm thickness) were exposed to Tris-buffered saline (TBS) containing 0.1% Triton X-100 and 2% Donkey serum (blocking buffer) and incubated overnight at 4 °C with primary antibodies in blocking buffer and then for 1–2 h at room temperature with Alexa Fluor-conjugated secondary antibodies in blocking buffer. For staining with the antibody to Cux1, Ctip2, Pax6, Tbr2 and Ki67, we performed antigen retrieval by autoclave treatment of sections with target retrieval solutions (Dako) for 10 min at 105 °C. Fluorescence images were obtained with a laser confocal microscope (Leica TCS-SP5). Antibodies used for immunostaining included Cux1 (Rabbit, 1:200, Santa Cruz, sc-13024), Ctip2 (Rat, 1:1000, Abcam, ab18465), NeuN (Mouse, 1:200, Millipore, MAB377), S100β (Rabbit, 1:500, Abcam, AB52642) Pax6 (Rabbit, 1:1000, Millipore, AB2237), Tbr2 (Chicken, 1:1000, Millipore, AB15894), Ki67 (Rat, 1:500, Dako, M7249) cleaved Caspase3 (Rabbit, 1:1000, Cell Signaling, 9664). Alexa-Fluor-labeled secondary antibodies (1:1000) and Hoechst were obtained from Life Technologies.
Western blotting
Total protein was extracted from the neocortex of P0 pups with the use of sonication. Lysates were prepared in 1 × Laemmli’s buffer and loaded in each well of a 10% acrylamide gel. Following transfer, the membranes were blocked with 2% skim milk in Tris-buffered saline containing 0.05% Tween20 and incubated overnight (4 °C) with the following primary antibodies: p57kip2(Cdkn1c) (Rabbit, 1:500, Sigma-Aldrich, P0357); β-actin (Mouse, 1:5000, Sigma-Aldrich, A2228). To examine a loading control, blots were stripped and reanalyzed for β-actin. The images were acquired by ImageQuant LAS4000 (GE Healthcare) and quantified using ImageQuant TL (GE Healthcare).
Northern blotting
One µg of total RNAs in a denaturing buffer were run on a 1% agarose gel, transferred and chemically crosslinked to Hybond-N (GE Healthcare). 28 S RNA was detected with methylene blue. After washing, membranes were hybridized with probes in DIG Easy Hyb Granules (Sigma Aldrich) at 65 °C. The region encoding Cdkn1c in the plasmid42 was transcribed with T3 promoter (antisense probe) and T7 promoter (sense probe), labeled with DIG RNA Labelling Mix (Roche) and used as probes. The images were acquired by ImageQuant LAS4000 (GE Healthcare) and quantified using ImageQuant TL (GE Healthcare).
Bisulfite analysis
Bisulfite treatment of the genomic DNA isolated from neocortex at E16 was performed using the EpiTect bisulfite kit (Qiagen). Each of KvDMR and ICG5 was amplified by PCR, and the products were subcloned and sequenced. The primer to amplify KvDMR1 (ICG8b) was previously described17. For ICG5, PCR amplification of the promoter region of Cdkn1c was carried out using primers below.
Fw GGAGTTGAAGGATTAGTTTTTTT
Rv ATATAAACATTTCCCCTTATCCC
RNA sequencing analysis
cDNA libraries were prepared using total RNA from neocortical tissues at E16 according to the Tru-seq library construction protocol. The library products were then sequenced by Illumina NovaSeq6000 (100 bp paired-end, stranded). Raw sequences were aligned to the mouse mm10 genome by HISAT243 using a transcriptome index built from RefSeq mm10 and visualized with the IGV browser. We tested for differential gene expression using edgeR and defined differentially expressed genes as having an adjusted p-value of less than 0.05. Functional annotation of the dysregulated genes (GO analysis) was performed with Metascape44. RNA-seq data is deposited at DDBJ (accession no. DRA009463).
Statistical analysis
Data are presented as means + s.e.m. as indicated, and were analysed by Student’s two-tailed paired t test and Student’s two-tailed unpaired t test, as indicated. A P value of <0.05 was considered statistically significant and the significance is marked by *P < 0.05 and **P < 0.01. The number of animals in each experiment is stated in the respective figure legends.
References
John, R. M. & Surani, M. A. Genomic imprinting, mammalian evolution, and the mystery of egg-laying mammals. Cell 101, 585–588, https://doi.org/10.1016/S0092-8674(00)80870-3 (2000).
Reik, W. & Walter, J. Evolution of imprinting mechanisms: The battle of the sexes begins in the zygote. Nat. Genet. 27, 255–256, https://doi.org/10.1038/85804 (2001).
Ferguson-Smith, A. C. Genomic imprinting: The emergence of an epigenetic paradigm. Nat. Rev. Genet. 12, 565–575, https://doi.org/10.1038/nrg3032 (2011).
Uribe-Lewis, S., Woodfine, K., Stojic, L. & Murrell, A. Molecular mechanisms of genomic imprinting and clinical implications for cancer. Expert Rev. Mol. Med. 13, 1–22, https://doi.org/10.1017/S1462399410001717 (2011).
Radford, E. J., Ferrón, S. R. & Ferguson-Smith, A. C. Genomic imprinting as an adaptative model of developmental plasticity. FEBS Lett. 585, 2059–2066, https://doi.org/10.1016/j.febslet.2011.05.063 (2011).
Peters, J. The role of genomic imprinting in biology and disease: An expanding view. Nat. Rev. Genet. 15, 517–530, https://doi.org/10.1038/nrg3766 (2014).
Demars, J. & Gicquel, C. Epigenetic and genetic disturbance of the imprinted 11p15 region in Beckwith-Wiedemann and Silver-Russell syndromes. Clin. Genet. 81, 350–361, https://doi.org/10.1111/j.1399-0004.2011.01822.x (2012).
Buiting, K. Prader-Willi syndrome and Angelman syndrome. Am. J. Med. Genet. Part C Semin. Med. Genet. 154, 365–376, https://doi.org/10.1002/ajmg.c.30273 (2010).
Crowley, J. J. et al. Analyses of allele-specific gene expression in highly divergent mouse crosses identifies pervasive allelic imbalance. Nat. Genet. 47, 353–360, https://doi.org/10.1038/ng.3222 (2015).
Ferrón, S. R. et al. Postnatal loss of Dlk1 imprinting in stem cells and niche astrocytes regulates neurogenesis. Nature 475, 381–385, https://doi.org/10.1038/nature10229 (2011).
Ferrón, S. R. et al. Differential genomic imprinting regulates paracrine and autocrine roles of IGF2 in mouse adult neurogenesis. Nat. Commun. 6, 8265, https://doi.org/10.1038/ncomms9265 (2015).
Hatada, I. & Mukai, T. Genomic imprinting of p57KIP2, a cyclin–dependent kinase inhibitor, in mouse. Nat. Genet. 11, 204–206, https://doi.org/10.1038/ng1095-204 (1995).
Matsuoka, S. et al. Imprinting encoding cyclin-dependent kinase inhibitor, p57KIP2,on chromosome 11p15. Genetics 93, 3026–3030 (1996).
Bhogal, B., Arnaudo, A., Dymkowski, A., Best, A. & Davis, T. L. Methylation at mouse Cdkn1c is acquired during postimplantation development and functions to maintain imprinted expression. Genomics 84, 961–970, https://doi.org/10.1016/j.ygeno.2004.08.004 (2004).
Fitzpatrick, G. V., Soloway, P. D. & Higgins, M. J. Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat. Genet. 32, 426–431, https://doi.org/10.1038/ng988 (2002).
Mancini-DiNardo, D., Steele, S. J. S., Levorse, J. M., Ingram, R. S. & Tilghman, S. M. Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev. 20, 1268–1282, https://doi.org/10.1101/gad.1416906 (2006).
Fan, T. et al. Lsh controls silencing of the imprinted Cdkn1c gene. Development 132, 635–644, https://doi.org/10.1242/dev.01612 (2005).
Lee, M. H., Reynisdttir, I. & Massague, J. Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev. 9, 639–649, https://doi.org/10.1101/gad.9.6.639 (1995).
Matsuoka, S. et al. p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 650–662, https://doi.org/10.1101/gad.9.6.650 (1995).
Zhang, P. et al. Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith–Wiedemann syndrome. Nature 387, 151–158, https://doi.org/10.1038/387151a0 (1997).
Westbury, J., Watkins, M., Ferguson-Smith, A. C. & Smith, J. Dynamic temporal and spatial regulation of the cdk inhibitor p57kip2 during embryo morphogenesis. Mech. Dev. 109, 83–89, https://doi.org/10.1016/S0925-4773(01)00512-3 (2001).
Hoovers, J. M. et al. Multiple genetic loci within 11p15 defined by Beckwith-Wiedemann syndrome rearrangement breakpoints and subchromosomal transferable fragments. Proc. Natl. Acad. Sci. USA 92, 12456–12460 (1995).
Hatada, I. et al. An imprinted gene p57KIP2 is mutated in Beckwith–Wiedemann syndrome. Genes Dev. 11, 973–983, https://doi.org/10.1038/ng1096-171 (1996).
Yan, Y., Frisén, J., Lee, M. H., Massagué, J. & Barbacid, M. Ablation of the CDK inhibitor p57 Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev. 11, 973–983, https://doi.org/10.1101/gad.11.8.973 (1997).
Takahashi, K. & Nakayama, K. Mice lacking a CDK inhibitor, p57Kip2, exhibit skeletal abnormalities and growth retardation. J. Biochem. 127, 73–83 (2000).
Itoh, Y., Masuyama, N., Nakayama, K., Nakayama, K. I. & Gotoh, Y. The cyclin-dependent kinase inhibitors p57 and p27 regulate neuronal migration in the developing mouse neocortex. J. Biol. Chem. 282, 390–396, https://doi.org/10.1074/jbc.M609944200 (2007).
Joseph, B. et al. p57(Kip2) cooperates with Nurr1 in developing dopamine cells. Proc. Natl. Acad. Sci. USA 100, 15619–15624, https://doi.org/10.1073/pnas.2635658100 (2003).
Joseph, B. et al. p57Kip2 is a repressor of Mash1 activity and neuronal differentiation in neural stem cells. Cell Death Differ. 16, 1256–1265, https://doi.org/10.1038/cdd.2009.72 (2009).
Tury, A., Mairet-Coello, G. & Dicicco-Bloom, E. The cyclin-dependent kinase inhibitor p57Kip2 regulates cell cycle exit, differentiation, and migration of embryonic cerebral cortical precursors. Cereb. Cortex 21, 1840–1856, https://doi.org/10.1093/cercor/bhq254 (2011).
Matsumoto, A. et al. Deregulation of the p57-E2F1-p53 Axis Results in Nonobstructive Hydrocephalus and Cerebellar Malformation in Mice. Mol. Cell. Biol. 31, 4176–4192, https://doi.org/10.1128/mcb.05370-11 (2011).
Furutachi, S., Matsumoto, A., Nakayama, K. I. & Gotoh, Y. P57 controls adult neural stem cell quiescence and modulates the pace of lifelong neurogenesis. EMBO J. 32, 970–981, https://doi.org/10.1038/emboj.2013.50 (2013).
Mairet-Coello, G. et al. p57KIP2 regulates radial glia and intermediate precursor cell cycle dynamics and lower layer neurogenesis in developing cerebral cortex. Development 139, 475–487, https://doi.org/10.1242/dev.067314 (2012).
Furutachi, S. et al. Slowly dividing neural progenitors are an embryonic origin of adult neural stem cells. Nat. Neurosci. 18, 657–665, https://doi.org/10.1038/nn.3989 (2015).
Van de Pette, M. et al. Visualizing Changes in Cdkn1c Expression Links Early-Life Adversity to Imprint Mis-regulation in Adults. Cell Rep. 18, 1090–1099, https://doi.org/10.1016/j.celrep.2017.01.010 (2017).
Bouschet, T. et al. In Vitro Corticogenesis from Embryonic Stem Cells Recapitulates the In Vivo Epigenetic Control of Imprinted Gene Expression. Cereb. Cortex 27, 2418–2433, https://doi.org/10.1093/cercor/bhw102. (2017).
Isaka, F. et al. Ectopic expression of the bHLH gene Math1 disturbs neural development. Eur. J. Neurosci. 11, 2582–2588, https://doi.org/10.1046/j.1460-9568.1999.00699.x (2003).
O’Neil, A. et al. Preventing mental health problems in offspring by targeting dietary intake of pregnant women. BMC Med. 12, 1–7, https://doi.org/10.1186/s12916-014-0208-0 (2014).
Vucetic, Z. et al. Early life protein restriction alters dopamine circuitry. Neuroscience 168, 359–370, https://doi.org/10.1016/j.neuroscience.2010.04.010 (2010).
McNamara, G. I., Davis, B. A., Dwyer, D. M., John, R. M. & Isles, A. R. Behavioural abnormalities in a novel mouse model for Silver Russell Syndrome. Hum. Mol. Genet. 25, 5407–5417, https://doi.org/10.1093/hmg/ddw357 (2016).
Mcnamara, G. I. et al. Dopaminergic and behavioural changes in a loss-of-imprinting model of Cdkn1c. Genes, Brain Behav. 149–157, https://doi.org/10.1111/gbb.12422 (2017).
Gaudet, M., Fara, A.-G., Beritognolo, I., & Sabatti, M. Allele-Specific PCR. In: Komar A. Single Nucleotide Polymorphisms. Methods in Molecular Biology™ (Methods and Protocols). 578. Humana Press, Totowa, NJ (2009)
Suzuki-Hirano, A. et al. Dynamic spatiotemporal gene expression in embryonic mouse thalamus. J. Comp. Neurol. 519, 528–543, https://doi.org/10.1002/cne.22531 (2011).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Zhou, Y. et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10 (2019).
Acknowledgements
We thank Toshihiko Shiroishi for providing us the genomic information of the JF1/Ms mouse strain; Keiichi Nakayama and Tsunaki Higa for providing Cdkn1c floxed mice; Ryoichiro Kageyama for providing Nestin-Cre mice; Yuki Katou and other members of Shirahige laboratory for help with sequencing analysis; Seth Blackshaw for providing the probe used for Northern blotting; and all members of the Gotoh laboratory for discussion and support. This study was supported by KAKENHI grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan and the Japan Society for the Promotion of Science (JP17J08651 for Y.I.; JP18K06477 for D.K.; JP16H06481, JP16H06279, JP16H06479, JP15H05773 for Y.G.) as well as by AMED-CREST of the Japan Agency for Medical Research and Development (JP19gm0610013 and JP19gm1310004), by the Uehara Memorial Foundation and by the International Research Center for Neurointelligence (WPI-IRCN) at The University of Tokyo Institutes for Advanced Study.
Author information
Authors and Affiliations
Contributions
Y.I. performed the experiments, analyzed the data, and wrote the manuscript. T.W. designed the allele specific qPCR of Cdkn1c. H.M. conducted initial analysis of Cdkn1c paternal cKO mice. D.K., S.F. and Y.G. conceived of and coordinated the project as well as wrote the manuscript. All authors approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Imaizumi, Y., Furutachi, S., Watanabe, T. et al. Role of the imprinted allele of the Cdkn1c gene in mouse neocortical development. Sci Rep 10, 1884 (2020). https://doi.org/10.1038/s41598-020-58629-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-58629-9
This article is cited by
-
Protein restriction during pregnancy alters Cdkn1c silencing, dopamine circuitry and offspring behaviour without changing expression of key neuronal marker genes
Scientific Reports (2024)
-
DNA Methylation Analysis of Imprinted Genes in the Cortex and Hippocampus of Cross-Fostered Mice Selectively Bred for Increased Voluntary Wheel-Running
Behavior Genetics (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
