
Abstract
We have surveyed 191 prospectively sampled familial cancer patients with no previously detected pathogenic variant in the BRCA1/2, PTEN, TP53 or DNA mismatch repair genes. In all, 138 breast cancer (BC) cases, 34 colorectal cancer (CRC) and 19 multiple early-onset cancers were included. A panel of 44 cancer-predisposing genes identified 5% (9/191) pathogenic or likely pathogenic variants and 87 variants of uncertain significance (VUS). Pathogenic or likely pathogenic variants were identified mostly in familial BC individuals (7/9) and were located in 5 genes: ATM (3), BRCA2 (1), CHEK2 (1), MSH6 (1) and MUTYH (1), followed by multiple early-onset (2/9) individuals, affecting the CHEK2 and ATM genes. Eleven of the 87 VUS were tested, and 4/11 were found to have an impact on splicing by using a minigene splicing assay. We here report for the first time the splicing anomalies using this assay for the variants ATM c.3806A > G and BUB1 c.677C > T, whereas CHEK1 c.61G > A did not result in any detectable splicing anomaly. Our study confirms the presence of pathogenic or likely pathogenic variants in genes that are not routinely tested in the context of the above-mentioned clinical phenotypes. Interestingly, more than half of the pathogenic germline variants were found in the moderately penetrant ATM and CHEK2 genes, where only truncating variants from these genes are recommended to be reported in clinical genetic testing practice.
Similar content being viewed by others


Introduction
Based on twin studies and recent reports, 30–45% of colorectal cancer (CRC) cases may involve a heritable component1,2,3,4. However, highly-penetrant pathogenic germline variants that explain familial aggregation and/or an early-onset of the disease (e.g. pathogenic variants in APC and DNA mismatch repair (MMR) genes) are detected in only 5–10% of the cases5. In contrast, in hereditary breast and ovarian cancer (HBOC), up to ~25% of the cases can be explained by the highly penetrant risk genes BRCA1 and BRCA26.
Next-generation sequencing (NGS) studies have reported that as many as ~18% of patients diagnosed with CRC < age of 50 years have pathogenic germline variants in genes that are not traditionally associated with CRC, including ATM, CHEK2, BRCA1, BRCA2, CDKN2A or PALB27,8, while up to 15% of HBOC patients harbor pathogenic variants in known BC predisposing genes, including RAD51C, RAD51D, ATM, CHEK2, BRIP1, PALB2, BARD1, RECQL, TP53, CDH1 and NBN9,10. Hence, in approximately 60%-70% of all tested hereditary CRC or HBOC patients, the genetic predisposition is still unknown. There is a need to determine whether these variants contribute to hereditary CRC or HBOC risk via the combination of low- and moderate-penetrance susceptibility alleles, or in conjunction with environmental factors7,8,11,12,13
Importantly, phenotype-driven panels provide a notable rate of detection of pathogenic variants with clinical actionability. NGS-based panel screening described 8–13% of pathogenic variants in BRCA1/2 negative HBOC patients14,15. Therefore, there are new genes recently discovered by the use of gene panel testing for which there is still limited data regarding the degree of their association to cancer risk, and the medical management guidelines are scant or non-existent for some of them16,17,18.
A relatively common event that complicates the interpretation of gene test results is the detection of variants of unknown significance (VUS). Regarding BRCA1 and BRCA2 genes, up to 20% of all variants are still being classified as VUS19, while about one-fifth to one-third are classified in the case of the DNA MMR gene variants20.
Because multiple cancer gene panel testing is rapidly replacing sequential single-gene testing, we need to know how to improve in the way we interpret the findings from panel testing patients in cancer kindreds who have been previously tested for the BRCA1/2, PTEN, TP53 and MMR genes without detection of pathogenic variants. The goals were to gain information on to which degree other genes may have been causative for cancer in the patients and their relatives, and to be informed on how such genes were deranged to discriminate between normal and disease-causing variants. In addition, we analyzed the impact of a subset of VUS on RNA splicing by the use of minigene assays.
Methods
The methods were performed in accordance with the relevant guidelines and regulations.
Study population
The study population was selected from the Hereditary Cancer Biobank (n = 161), which is part of the out-patient inherited cancer clinic from the Norwegian Radium Hospital (Norway)21 and the Department of Genomic Medicine (n = 30) from the University of Manchester (United Kingdom)12.
The selection criteria for the 191 individuals were as follows:
-
Being member of cancer family or having a personal history of cancer;
-
Presence of multiple early-onset cancer at early age of onset, including BC, gynecological cancer, CRC, or thyroid cancer;
-
Familial CRC cases: Families that fulfilled the Amsterdam criteria or the revised Bethesda guidelines who had tested negative for pathogenic variants in the mismatch repair (MMR) genes;
-
Familial BC cases: Women with BC or gynecological cancer who had tested negative for pathogenic BRCA1 or BRCA2 variants.
Overall, our study subjects (n = 191) were demonstrated not to carry pathogenic variants in BRCA1/2, TP53, PTEN or MMR genes, or large exon deletions/duplications involving these genes by standard diagnostics.
In all, 138 breast cancer (BC) cases were included, where 57 were denoted as BRCA phenocopies (cases who were tested negative for their respective family’s path_BRCA1 (n = 18) and path_BRCA2 variants (n = 39)). Thirty-four CRC and 19 multiple early-onset cancers cases were also included (Fig. 1). Out of the 191 familial cancer patients, we have previously reported on 95 individuals (48 phenocopies, 34 familial CRC and 13 multiple early-onset cases)12,22,23. The current study added 96 new cases, including 81 familial BC cases, 9 phenocopies and 6 additional subjects having multiple early-onset cancer (Fig. 1).

Relational flowchart of the cancer kindreds and results from the study.
Ethical approval for the study was granted by the Norwegian Data Inspectorate (ref. 2001/2988-2) and Ethical Review Board (ref. S-02030). All examined patients signed an informed consent for their participation in the study.
Targeted sequencing and data analysis
Genomic DNA was isolated from peripheral blood samples and a 44-gene panel targeted sequencing was carried out using a TruSeq amplicon-based assay v.1.5 on a MiSeq apparatus (Supplementary Table 1)12,22,23. The mean depth of sequencing coverage ranged from 124 to 535, and the fraction of target bases with coverage ≥25 ranged from 80% to 94%.
Sequence alignments with paired-end sequence reads to the human reference genome (build GRCh37) were obtained through the BWA-mem algorithm (v.0.7.8-r55), where sorting and indexing procedures were undertaken with Samtools (v.1.1). Genotyping of single nucleotide variants (SNV) and short indels, as well as quality filtering of the raw genotype calls, were done according to GATK’s best practice procedures, as described previously in more detail12,22,23.
Functional annotation of the variants was performed with ANNOVAR (version November 2015)24 and using five databases (gnomAD, dbSNP (build 147)25, 1000 Genome Project phase326, UniProt (release March 2016)27, and Pfam protein domain (v29, December 2015)12,22,23,28.
Nomenclature and classification of genetic variants
The Human Genome Variation Society (HGVS) guidelines were used to describe the detected genetic variants29. The following databases: Evidence-based Network for the Interpretation of Germline Mutant Alleles (ENIGMA), Breast Cancer Information Core (BIC), International Society of Gastrointestinal Hereditary Tumors (InSiGHT), Leiden Open Variation Database (LOVD), ClinVar, and the Human Gene Mutation Database (HGMD) were interrogated for the clinical significance of the identified genetic variants (in their releases as of August 2018).
We applied the American College of Medical Genetics and Genomics (ACMG) guidelines for the classification and interpretation of the clinical significance of the variants30. In addition, when a novel variant was identified in the study, we considered it to be pathogenic or likely pathogenic if either one of the following criteria were met: a) introduction of a premature stop codon in the protein coding sequence (nonsense or frameshift); b) occurrence at positions +1/+ 2 or −1/−2 of donor or acceptor splice sites, respectively; or c) whole-exon deletions.
Validation by cycling temperature capillary electrophoresis
Cycling temperature capillary electrophoresis was used to validate the 13 pathogenic variants found in the study. The method has been previously described12,31,32,33. The variant melting profile tool (http://meltprimer.ous-research.no/)34 was used to design the amplicons. Details about the primer sequences, PCR reaction conditions and electrophoresis settings are available upon request.
In silico analyses of VUS
The MaxEntScan (MES) and SSF-like (SSFL) methods were used to predict variant-induced alterations in 3′ and 5′ splice site (3′ss and 5′ss) strength, as described by Houdayer et al. 201235, but using the integrated software tool Alamut Batch version 1.5 (Interactive Biosoftware, France). As previously described by Soukarieh et al.36 and in prior studies12,22,23, we considered:
-
If ΔMES ≥ 15% and ΔSSFL ≥ 5%: the variant mapping at a splice site could negatively impact exon inclusion35;
-
If negative Δ scores were provided by at least 2 out of the 3 exonic splicing regulatory elements (ESR)-dedicated in silico tools: the variant located outside the splice sites was considered as a probable inducer of exon skipping36.
The Align-GVGD (the VUS were predicted as deleterious when the values were C35 or higher), SIFT, MAPP, PolyPhen-2 and MutationTaster12,22,23,37,38,39,40,41 were used to predict the protein impact of missense variants. The Alamut Batch version 1.4.4 (Interactive Biosoftware, http://www.interactive-biosoftware.com) was used for the protein in silico predictions.
Cell-based minigene splicing assay
We performed functional assays based on comparative analysis of the splicing pattern of wild type (WT) and mutant reporter minigenes to analyze the impact on splicing of each selected variant, as previously reported12,22,23.
The genomic regions containing the exon of interest and ~150 nucleotides of the flanking introns were amplified by PCR using patients’ DNA as template and primers (Supplementary Table 2). The PCR-amplified fragments were inserted into a previously linearized pCAS2 vector, generating the representative minigenes. The inserts of all clones were sequenced and the WT and mutant constructs were transfected in parallel into HeLa cells grown at ~70% confluence. The total RNA was extracted after 24 h, and the transcripts were analyzed by semi-quantitative RT-PCR (Supplementary Table 2). The RT-PCR products were separated by electrophoresis, followed by gel-purification. Sanger sequencing was performed for the proper identification of the minigene transcripts. Finally, fluorescent RT-PCR reactions followed by capillary electrophoresis were performed to quantify the splicing events. The computational analysis was performed using GeneMapper v5.0 software (Applied Biosystems).
Immunohistochemistry of ATM c.3806A > G carrier
We assessed in situ the effect of ATM c.3806A > G p.(Lys1269Arg) at the protein level by performing an immunohistochemistry (IHC) analysis. Paraffin-embedded material from the patient tumor was immuno-stained for the ATM protein: Briefly, 4-µm sections were placed on SuperFrost® Plus microscope slides. Antigen retrieval was performed in a Dako PT Link in Dako’s pH 6 (Low pH) retrieval solution according to the Dako’s Flex protocol and stained manually. We used the anti-ATM antibody clone 2C1 (Abcam ab78, dilution 1:2000). The ATM protein expression was assessed either as retained (normal), absent, or weak (i.e. tumor cell staining intensity was reduced compared with that of the normal internal control).
Results
Pathogenic or likely pathogenic germline findings
We utilized a 44-cancer gene panel in 191 high-risk individuals in order to identify other high- or moderate- penetrance genes that could be implicated in the genetic cause of these phenotypes (Supplementary Table 1). In the combined cohort, we identified 9 cases (5% of 191) who carried 9 pathogenic or likely pathogenic variants that were confirmed by cycling temperature capillary electrophoresis, showing 100% concordance (Fig. 1, Table 1).
From high- to moderate-penetrance genes identified in the study
Two pathogenic variants (2/191, 1%) were identified in high-penetrance genes, namely BRCA2 c.9382C > T p.(Arg3128*) (this variant was previously undetected, as only the path_BRCA2 c.6591_6592delTG was demonstrated in a relative subjected to previous testing) and MSH6 c.3261delC (p.Phe1088Serfs*2) from two phenocopy individuals.
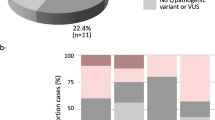
In the case of the heterozygous MUTYH c.1178G > A (p.Gly393Asp), we reported this gene as a recessive risk and it was found in a phenocopy individual, as previously reported by us12 (Table 1, Fig. 2A).

Overview of the germline variants found in the study. (A) Type of the pathogenic or likely pathogenic variants found in 191 multiple early-onset and familial cancers from multigene NGS. (B). Distribution of pathogenic or likely pathogenic variants by clinical phenotype
Six pathogenic variants (6/191, 3.1%) were detected in moderate-penetrance genes and included: ATM c.9139C > T (p.Arg3047*) and ATM c.468G > A (p.Trp156*) found in two phenocopies cases12, the Norwegian founder ATM c.3245_3247delinsTGAT (p.His1082Leufs)42, and the ATM c.8432delA (p.Lys2811Serfs*46) found in familial BC and multiple early-onset case, respectively. In addition, we identified CHEK2 c.319 + 2T > A (IVS2 + 2T > A in multiple early-onset and familial BC cases (Fig. 2A).
Pathogenic or likely pathogenic variants by clinical phenotype
When analyzed by clinical phenotype, we described that phenocopies and familial BC patients are the most affected cohorts by the presence of pathogenic variants in high- and moderate-penetrance genes, respectively. In detail, phenocopies were affected by the presence of pathogenic variants in the high-penetrance genes BRCA2 and MSH6, the moderate-penetrance gene: ATM and MUTYH gene. Familial BC cases were predominately affected by pathogenic or likely pathogenic variants in moderate-penetrance (ATM, CHEK2) genes (Fig. 2B).
In silico and minigene analysis of VUS
In addition to the 9 pathogenic variants found in the study, the NGS results revealed the presence of 87 variants that we interpreted as VUS. When in silico splicing and protein predictions were applied, 17 VUS in 15 genes were selected as more likely to be functionally relevant: APC, ATM, AXIN2, BRCA2, BUB1, CDH1, CHEK1, CHEK2, MAP3K1, MSH2, NOTCH3, POLE, RAD51B, RAD51D, and STK11 (Supplementary Table 3). The MAF of these variants in the gnomAD database were very low (<1%) or not reported (Supplementary Table 3). Given that patient RNA was not available, we decided to experimentally assess the impact on splicing of the 17 VUS by performing cell-based minigene assays. Because of technical difficulties, 6 variants were excluded from the analysis (Supplementary Table 3).
An overview of the results for the remaining 11 VUS is shown in Supplementary Table 3, which includes data for 8 variants that we have published elsewhere. More precisely, we previously reported that APC c.721G > A, BRCA2 c.116C > T, CDH1 c.1774G > T, CHEK2 c.608A > G MAP3K1 c.764A > G and NOTCH3 c.5854G > A did not affect the splicing pattern of the minigene transcripts12,22, whereas MSH2 c.815C > T and NOTCH3 c.1490C > T were previously described to induce partial skipping of exons 5 and 9, respectively22,43. Here, we report that ATM c.3806A > G induces a complex pattern of aberrant splicing as described below (Fig. 3A–D). Moreover, we found that BUB1 c.677C > T causes a moderate increase in exon skipping, whereas CHEK1 c.61G > A has no major effect in the context of our minigene assay (Fig. 3B).

Detection of variant-induced splicing alterations by using a minigene splicing assay. (A) Structure of pCAS2 minigenes used in this assay. The gray arrow indicates the CMV promoter, boxes represent exons, lines in between indicate introns, and arrows below the exons represent primers used in RT-PCR reactions. The start indicates a fluorescent primer. (B) Analysis of the splicing pattern of the pCAS2-derived minigenes carrying ATM, BUB1 and CHEK1 variants as indicated. The minigene assays were performed as described under Materials and Methods. The top panel shows the RT-PCR products obtained for WT and mutant constructs separated by electrophoresis on an ethidium bromide–stained 2.5% agarose gel. The bottom panel shows the quantification of the RT-PCR products focusing on the relative level of exon inclusion (i.e. products corresponding to full-length minigene transcripts, FL). Quantification was performed after separating the fluorescent RT-PCR products by capillary electrophoresis on an automated sequencer. as described under Materials and Methods. (C) Representative fluorescent RT-PCR experiment using pCAS2 minigenes carrying ATM exon 26 (WT and c.3806A > G, as indicated). The panel shows superposed peaks corresponding to the WT and mutant RT-PCR products (in blue and red, respectively). Splicing events are expressed as % of the total amount of RT-PCR products obtained for each WT and mutant minigene construct (mutant vs WT). Results are representative of two independent experiments. (D) Splice sites-dedicated bioinformatics predictions and representation of the major splicing events detected in the minigene assay relative to ATM exon 26. The position of c.3806A > G is indicated by the red circle. In silico predictions of splice sites were obtained by simultaneously interrogating 5 algorithms (SpliceSiteFinder-Like, MaxEntScan, NNSPLICE, GeneSplicer and Human Splice Finder) through the integrated software Alamut Visual Version 2.10 (Interactive Biosoftware, France). For simplicity, only scores relative to ATM 5′ss and 3′ss (natural, de novo or cryptic sites) detected in the minigene assay are shown. The properly spliced full-length exon (FL) is represented above the panel whereas the aberrantly spliced truncated versions of the exon are indicated underneath. (E) Immunohistochemical staining for ATM in the patient’s tumor. (F) Breast carcinoma control. (G) Malignant effusions from other cancer cases. Host cells, consisting of lymphocytes, macrophages, stromal cells and adipocytes, are strongly positive. The tumor was immunostained for the ATM protein as follows. Briefly, 4-µm sections were placed on SuperFrost® Plus microscope slides. Antigen retrieval was performed in a Dako PT Link in Dako’s pH 6 (Low pH) retrieval solution according to the Dako´s Flex protocol and stained manually. We used the anti-ATM antibody clone 2C1 (Abcam ab78, dilution 1:2000). The ATM protein expression was assessed as retained (normal), absent, or weak (i.e. tumor cell staining intensity was reduced compared with that of the normal internal control).
ATM c.3806A > G, BUB1 c.677C > T and CHEK1 c.61G > A
In the ATM gene, we identified a transition A > G at position c.3806 in the exon 26 (c.3806A > G, p.(Lys1269Arg), rs146017595) in a woman with a BC diagnosis at 46 years. The pedigree indicated a family history of BC, ovarian cancer and colon cancer, including a mother with BC diagnosis at 56 years, and two maternal aunts with BC diagnoses at 43 and 46 years, respectively (data not shown). To our knowledge, ATM c.3806A > G (p.Lys1269Arg) has not been classified in the literature either as pathogenic or benign. The variant is present in the general population at a very low allele frequency according to the gnomAD database (6.668 e-05) and is reported in ClinVar as a VUS (Variation ID: 185981). RNA splicing-dedicated in silico analyses predicted that this variant may affect RNA maturation by modifying potential splicing signals within ATM exon 26 (Supplementary Table 3). Our minigene assay results, shown in Fig. 3, revealed that ATM c.3806A > G indeed causes aberrant splicing, leading to a decrease in full-length transcripts (from 95% to 23%). Two main aberrant isoforms were detected in this assay: one presenting a terminal deletion of 192 nucleotides (corresponding to ATM r.3802_3993del, p.Val1268_Glu1331del) and the other presenting an internal deletion of 102 nucleotides within ATM exon 26 (corresponding to ATM r.3803_3904del, p.Val1268_1301del). Both alterations result from the usage of a new splice site directly introduced by the variant as predicted by the MaxEntScan algorithm (Supplementary Table 3). The terminal deletion indicates that the new 5′ ss can, in some transcripts, outcompete the natural 5′ss of exon 26 and be spliced to the following exon, whereas the internal deletion shows that ATM c.3806A > G also leads, in part, to the activation of a cryptic 3′ss downstream in exon 26 (Fig. 3D). Protein-based in silico analyses were inconsistent regarding the effect that ATM c.3806A > G (p.Lys1269Arg) may have on protein structure and function (Supplementary Table 3). We assessed in situ the effect at the protein level of ATM c.3806A > G by IHC analysis. The results revealed a weak-to-absent staining in the proband’s tumor as compared to normal surrounding tissue (Fig. 3E–G), also suggesting that the germline ATM c.3806A > G may be implicated in tumor development.
The BUB1 c.677C > T variant was identified in a female with a BC diagnosis at 56 years who did not carry any pathogenic variant in BRCA1/2 or MMR genes. In addition to the BUB1 variant, the patient carried two class 1/2 variants: NOTCH3 c.1487C > T (p.Pro496Leu) and NOTCH3 c.3704A > T (p.His1235Leu). The BUB1 variant is present in the general population at a low allele frequency according to the gnomAD database (1.999 e-3), and has not been reported in ClinVar. As ATM c.3806A > G, the protein-based in silico analyses were inconsistent regarding the effect on protein structure and function for BUB1 c.677C > T (Supplementary Table 3). The minigene assay showed a moderate increase in exon skipping (43% to 61%) (Fig. 3B).
Finally, we identified a CHEK1 c.61G > A in a BRCA2 phenocopy case, which is currently not reported in ClinVar, and the minigene assay showed no major splicing defect, which is in contrast to the in silico predictions (Fig. 3B).
Discussion
When applying multigene panel testing to a set of 191 familial cancer cases, we identified 9 cases (5%) harboring pathogenic or likely pathogenic variants. Most of these variants (4.2%, 8/191) were identified in the moderate to high penetrance genes BRCA2, MSH6, CHEK2 and ATM. The detection rate in these core genes is comparable to previous reports14,15. We also detected a likely pathogenic variant in MUTYH, for which there is still insufficient evidence for a significant risk of BC.
Interpreting the pathogenicity significance of moderate- or low- penetrance variants is a challenge for current cancer risk management, counselling and treatment decision-making regarding patients and their families. In this regard, Lynch syndrome (LS) is an autosomal dominant condition caused by pathogenic variants in one of the MMR genes, which result in different cancer risks. Path_MLH1 and path_MSH2 variants are highly penetrant when comparing to path_MSH6 and path_PMS2 variants44,45,46. Regarding BRCA genes, we have recently described that the most frequent path_BRCA1 variants in the Norwegian population had low penetrance in fertile ages, and lower than in carriers of infrequent path_BRCA1 variants47. The relative risk of developing BC has been described for truncating variants in BRCA2 (relative risk: 11.7), while ATM and CHEK2 truncating variants have relative risks of 2.8 (2.2–3.7) and 3.0 (2.6–3.5), respectively48.
A recent evaluation of gene-disease associations using the ClinGen clinical validity framework revealed definitive assertion for ATM and CHEK2 genes in breast cancer49. Our results are in agreement for the pathogenic variants found in the ATM (n = 3) and CHEK2 (n = 1) genes which were found in phenocopies or familial BC cases49.
There is still no clear evidence of an association between BC and other DNA repair genes, such as MLH1, MSH2, MSH6 and MAP3K1 genes. An average moderate-risk colorectal screening based on family history has been suggested for individuals with a heterozygous germline MUTYH pathogenic variant50,51.
Approximately 25% of missense and nonsense variants disrupt mRNA splicing, and the understanding of functional splice variations may allow the (re)classification of VUS in CRC and other BC-associated genes. In a recent retrospective study, among variants initially classified as VUS, 7.7% were reclassified (91% were downgraded to class 1/2 and 9% were upgraded class 4/5) and 8.7% were upgraded to class 4/552.
Out of the 87 variants of VUS, 11 were tested by a minigene splicing assay, out of which two (ATM c.3806A > G and BUB1 c.677C > T) were demonstrated to exert an effect on splicing. The ATM c.3806A > G variant was identified in an early-onset BC patient (<50 years) with a strong BC, ovarian and colon cancer family history. Our minigene assay results suggested that this variant partially alters the splicing pattern of ATM, leading to the concomitant production of aberrant transcripts presenting in-frame deletions in exon 26 and a decreased expression of full-length transcripts carrying the c.3806A > G (p.Lys1269Arg) variant. Although these experimental findings agree with RNA-splicing-dedicated in silico predictions, and minigene results are often concordant with in vivo data43,53,54, it will be essential to confirm our experimental results by analyzing RNA from patient samples. However, no RNA from the peripheral blood nor from the tumor of this patient, nor the other patients, were available for analysis. Our IHC results suggest that ATM c.3806A > G is associated with tumor development in the index patient. However, segregation data may provide additional evidence to classify this variant.
There is no clinical significance reported for the BUB1 c.677C > T variant. BUB1 encodes a serine/threonine-protein kinase that plays a central role in mitosis55, and pathogenic variants in this gene have been associated with aneuploidy and several forms of cancer. However, a recent study does not support the inclusion of BUB1 and BUB3 testing in routine genetic diagnostics of familial CRC and polyposis56. Our minigene assay showed a moderate increase in exon skipping due to the BUB1 c.677C > T variant, that remains classified as a VUS.
Information about ATM c.3806A > G and BUB1 c.677C > T variants in the spectrum of BC studies using NGS panel testing is currently scarce. In this scenario, not only functional or protein assays, but also co-segregation studies, will be essential for understanding whether the VUS analyzed in this work have causal or modifying effects, or otherwise are non-pathogenic. Importantly, there is no systematic classification for most of the genetic variants found by NGS, including the ATM c.3806A > G and BUB1 c.677C > T identified in this study, and, in more general terms, the impact of pathogenic variants in low- to moderate-penetrance genes is not fully understood with respect to clinical management. Most of the VUS may in the future be reclassified as deleterious or benign, but in the meantime, they cannot be used to make clinical decisions22.
There is a need to understand and quantify the cancer risk of pathogenic and currently uncertain variants found in moderate- or low- penetrance genes. A quantitative approach to prioritize missense substitutions with high probabilities of pathogenicity has been suggested for the ATM, CHEK2 and NBN genes57. However, there is still a high number of novel variants with an unknown significance of pathogenicity identified in cancer patients with family history awaiting classification. The evaluation of clinical, family parameters, functional assay and IHC analyses undertaken in this study may allow to refine the clinical classification of these variants. To the latter end, broad collaborations to compile data from different sources are needed to arrive at conclusions, and, we here contribute our results towards that end.
Conclusion
Five percent of 191 families initially tested by clinical diagnostic settings, were found to carry pathogenic variants in the ATM, MSH6, BRCA2, MUTYH and CHEK2 genes, when using a 44-gene panel. We further provided functional testing and protein characterization in addition to clinical and family history information.
Our study contributes to understanding and interpreting variants in moderate- or low-penetrance genes which are not routinely tested in the clinical genetic settings and may have a direct impact on providing an appropriate genetic counseling and clinical management for individuals and their relatives who carry these variants.
References
Mucci, L. A. et al. Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 315, 68–76, https://doi.org/10.1001/jama.2015.17703 (2016).
Grady, W. M. Genetic testing for high-risk colon cancer patients. Gastroenterology 124, 1574–1594 (2003).
Lichtenstein, P. et al. Environmental and heritable factors in the causation of cancer–analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 343, 78–85, https://doi.org/10.1056/NEJM200007133430201 (2000).
AlDubayan, S. H. et al. Inherited DNA-Repair Defects in Colorectal Cancer. Am J Hum Genet 102, 401–414, https://doi.org/10.1016/j.ajhg.2018.01.018 (2018).
Hahn, M. M. et al. The genetic heterogeneity of colorectal cancer predisposition - guidelines for gene discovery. Cell Oncol (Dordr) 39, 491–510, https://doi.org/10.1007/s13402-016-0284-6 (2016).
Kast, K., Dobberschütz, C., Sadowski, C. E., Pistorius, S. & Wimberger, P. Prevalence of Lynch syndrome in unselected patients with endometrial or ovarian cancer. Archives of gynecology and obstetrics 294, 1299–1303 (2016).
Pearlman, R. et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol, https://doi.org/10.1001/jamaoncol.2016.5194 (2016).
Kastrinos, F. et al. Development and Validation of the PREMM5 Model for Comprehensive Risk Assessment of Lynch Syndrome. J Clin Oncol 35, 2165–2172, https://doi.org/10.1200/JCO.2016.69.6120 (2017).
Ramus, S. J. et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J Natl Cancer Inst 107, https://doi.org/10.1093/jnci/djv214 (2015).
Gupta, S. et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 3.2017. J Natl Compr Canc Netw 15, 1465–1475, https://doi.org/10.6004/jnccn.2017.0176 (2017).
Picelli, S. et al. Meta-analysis of mismatch repair polymorphisms within the cogent consortium for colorectal cancer susceptibility. Plos One 8, e72091, https://doi.org/10.1371/journal.pone.0072091 (2013).
Dominguez-Valentin, M. et al. Genetic variants of prospectively demonstrated phenocopies in BRCA1/2 kindreds. Hered Cancer Clin Pract 16, 4, https://doi.org/10.1186/s13053-018-0086-0 (2018).
Boland, P. M., Yurgelun, M. B. & Boland, C. R. Recent progress in Lynch syndrome and other familial colorectal cancer syndromes. CA Cancer J Clin. https://doi.org/10.3322/caac.21448 (2018).
Schubert, S. et al. The identification of pathogenic variants in BRCA1/2 negative, high risk, hereditary breast and/or ovarian cancer patients: High frequency of FANCM pathogenic variants. Int J Cancer, https://doi.org/10.1002/ijc.31992 (2018).
Tedaldi, G. et al. Multiple-gene panel analysis in a case series of 255 women with hereditary breast and ovarian cancer. Oncotarget 8, 47064–47075, https://doi.org/10.18632/oncotarget.16791 (2017).
Taylor, A. et al. Consensus for genes to be included on cancer panel tests offered by UK genetics services: guidelines of the UK Cancer Genetics Group. Journal of medical genetics, jmedgenet-2017-105188 (2018).
Anele, C. C. et al. Risk of metachronous colorectal cancer following colectomy in Lynch syndrome: a systematic review and meta-analysis. Colorectal Dis 19, 528–536, https://doi.org/10.1111/codi.13679 (2017).
Huang, K. L. et al. Pathogenic Germline Variants in 10,389 Adult Cancers. Cell 173, 355–370 e314, https://doi.org/10.1016/j.cell.2018.03.039 (2018).
Eccles, D. M. et al. BRCA1 and BRCA2 genetic testing-pitfalls and recommendations for managing variants of uncertain clinical significance. Ann Oncol 26, 2057–2065, https://doi.org/10.1093/annonc/mdv278 (2015).
Plazzer, J. P. et al. The InSiGHT database: utilizing 100 years of insights into Lynch Syndrome. Familial Cancer 12, 175–180, https://doi.org/10.1007/s10689-013-9616-0 (2013).
Crosbie, E. J. et al. The Manchester International Consensus Group recommendations for the management of gynecological cancers in Lynch syndrome. Genet Med, https://doi.org/10.1038/s41436-019-0489-y (2019).
Dominguez-Valentin, M. et al. Potentially pathogenic germline CHEK2 c.319 + 2T > A among multiple early-onset cancer families. Fam Cancer, https://doi.org/10.1007/s10689-017-0011-0 (2017).
Dominguez-Valentin, M. et al. Identification of genetic variants for clinical management of familial colorectal tumors. BMC medical genetics 19, 26, https://doi.org/10.1186/s12881-018-0533-9 (2018).
Auclair, J. et al. Intensity-dependent constitutional MLH1 promoter methylation leads to early onset of colorectal cancer by affecting both alleles. Genes Chromosomes Cancer 50, 178–185, https://doi.org/10.1002/gcc.20842 (2011).
Sherry, S. T. et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29, 308–311 (2001).
Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68–74, https://doi.org/10.1038/nature15393 (2015).
Apweiler, R. et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res 32, D115–D119, https://doi.org/10.1093/nar/gkh131 (2004).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res 42, D222–D230, https://doi.org/10.1093/nar/gkt1223 (2014).
den Dunnen, J. T. & Antonarakis, S. E. Mutation nomenclature extensions and suggestions to describe complex mutations: A discussion. Hum Mutat 15, 7–12 (2000).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424, https://doi.org/10.1038/gim.2015.30 (2015).
Ekstrom, P. O., Khrapko, K., Li-Sucholeiki, X. C., Hunter, I. W. & Thilly, W. G. Analysis of mutational spectra by denaturing capillary electrophoresis. Nat Protoc 3, 1153–1166, https://doi.org/10.1038/nprot.2008.79 (2008).
Ekstrom, P. O., Warren, D. J. & Thilly, W. G. Separation principles of cycling temperature capillary electrophoresis. Electrophoresis 33, 1162–1168, https://doi.org/10.1002/elps.201100550 (2012).
Hinselwood, D. C., Abrahamsen, T. W. & Ekstrom, P. O. BRAF mutation detection and identification by cycling temperature capillary electrophoresis. Electrophoresis 26, 2553–2561, https://doi.org/10.1002/elps.200410427 (2005).
Ekstrom, P. O., Nakken, S., Johansen, M. & Hovig, E. Automated amplicon design suitable for analysis of DNA variants by melting techniques. BMC Res Notes 8, 667, https://doi.org/10.1186/s13104-015-1624-8 (2015).
Houdayer, C. et al. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum Mutat 33, 1228–1238, https://doi.org/10.1002/humu.22101 (2012).
Soukarieh, O. et al. Exonic Splicing Mutations Are More Prevalent than Currently Estimated and Can Be Predicted by Using In Silico Tools. Plos Genet 12, https://doi.org/10.1371/journal.pgen.1005756 (2016).
Adzhubei, I. A. et al. A method and server for predicting damaging missense mutations. Nat Methods 7, 248–249, https://doi.org/10.1038/nmeth0410-248 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4, 1073–1082, https://doi.org/10.1038/nprot.2009.86 (2009).
Schwarz, J. M., Cooper, D. N., Schuelke, M. & Seelow, D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods 11, 361–362, https://doi.org/10.1038/nmeth.2890 (2014).
Stone, E. A. & Sidow, A. Physicochemical constraint violation by missense substitutions mediates impairment of protein function and disease severity. Genome Res 15, 978–986, https://doi.org/10.1101/gr.3804205 (2005).
Tavtigian, S. V. et al. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. Journal of Medical Genetics 43, 295–305, https://doi.org/10.1136/jmg.2005.033878 (2006).
Borresen, A. L., Andersen, T. I., Tretli, S., Heiberg, A. & Moller, P. Breast cancer and other cancers in Norwegian families with ataxia-telangiectasia. Genes Chromosomes Cancer 2, 339–340 (1990).
Tournier, I. et al. A Large Fraction of Unclassified Variants of the Mismatch Repair Genes MLH1 and MSH2 Is Associated With Splicing Defects. Hum Mutat 29, 1412–1424, https://doi.org/10.1002/humu.20796 (2008).
Dominguez-Valentin, M. et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med, https://doi.org/10.1038/s41436-019-0596-9 (2019).
Moller, P. et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut, https://doi.org/10.1136/gutjnl-2015-309675 (2015).
Moller, P. et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database. Gut, https://doi.org/10.1136/gutjnl-2017-314057 (2017).
Moller, P., Dominguez-Valentin, M., Rodland, E. A. & Hovig, E. Causes for Frequent Pathogenic BRCA1 Variants Include Low Penetrance in Fertile Ages, Recurrent De-Novo Mutations and Genetic Drift. Cancers (Basel) 11, https://doi.org/10.3390/cancers11020132 (2019).
Easton, D. F. et al. Gene-panel sequencing and the prediction of breast-cancer risk. N Engl J Med 372, 2243–2257, https://doi.org/10.1056/NEJMsr1501341 (2015).
Lee, K. et al. Clinical validity assessment of genes frequently tested on hereditary breast and ovarian cancer susceptibility sequencing panels. Genet Med. https://doi.org/10.1038/s41436-018-0361-5 (2018).
Nielsen, M., Lynch, H., Infante, E. & Brand, R. In GeneReviews((R)) (eds M. P. Adam et al.) (1993).
Out, A. A. et al. MUTYH gene variants and breast cancer in a Dutch case-control study. Breast Cancer Res Treat 134, 219–227, https://doi.org/10.1007/s10549-012-1965-0 (2012).
Mersch, J. et al. Prevalence of Variant Reclassification Following Hereditary Cancer Genetic Testing. JAMA 320, 1266–1274, https://doi.org/10.1001/jama.2018.13152 (2018).
Auclair, J. et al. Systematic mRNA analysis for the effect of MLH1 and MSH2 missense and silent mutations on aberrant splicing. Hum Mutat 27, 145–154, https://doi.org/10.1002/humu.20280 (2006).
van der Klift, H. M. et al. Splicing analysis for exonic and intronic mismatch repair gene variants associated with Lynch syndrome confirms high concordance between minigene assays and patient RNA analyses. Mol Genet Genomic Med 3, 327–345, https://doi.org/10.1002/mgg3.145 (2015).
Raaijmakers, J. A. et al. BUB1 Is Essential for the Viability of Human Cells in which the Spindle Assembly Checkpoint Is Compromised. Cell Rep 22, 1424–1438, https://doi.org/10.1016/j.celrep.2018.01.034 (2018).
Bubien, V. et al. Combined tumor genomic profiling and exome sequencing in a breast cancer family implicates ATM in tumorigenesis: A proof of principle study. Genes Chromosomes Cancer, https://doi.org/10.1002/gcc.22482 (2017).
Young, E. L. et al. Multigene testing of moderate-risk genes: be mindful of the missense. J Med Genet 53, 366–376, https://doi.org/10.1136/jmedgenet-2015-103398 (2016).
Acknowledgements
The authors wish to thank Karen-Marie Heintz and Geir Frode Øy for the excellent experimental assistance. This work was supported by the Radium Hospital Foundation (Oslo, Norway), Helse Sør-Øst (Norway), the Norwegian Cancer Society, contract 194751-2017, the Groupement des Entreprises Françaises dans la Lutte contre le Cancer (Gefluc), the Association Nationale de la Recherche et de la Technologie (ANRT, CIFRE PhD fellowship to H.T.) and by the OpenHealth Institute. This study was also cosupported by Inserm, European Union and Région Normandie. Europe gets involved in Normandie with European Regional Development Fund (ERDF).
Author information
Authors and Affiliations
Contributions
All authors have taken part in the different steps of the study: M.D.V., P.M., D.G.E., G.C. and E.H. designed the study, A.M., H.T. performed in silico splicing predictions and the minigene assays, P.O.E. performed validation experiments, M.M., A.N. and E.H.F. performed in silico protein predictions, S.N., D.V. performed the sequence analysis, A.H., B.D. performed immunochemistry analysis and evaluation. M.D.V. drafted the manuscript and all have read, revised and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dominguez-Valentin, M., Nakken, S., Tubeuf, H. et al. Results of multigene panel testing in familial cancer cases without genetic cause demonstrated by single gene testing. Sci Rep 9, 18555 (2019). https://doi.org/10.1038/s41598-019-54517-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-54517-z
This article is cited by
-
Outcomes of retesting in patients with previously uninformative cancer genetics evaluations
Familial Cancer (2022)
-
Prevalence of germline variants in consensus moderate-to-high-risk predisposition genes to hereditary breast and ovarian cancer in BRCA1/2-negative Brazilian patients
Breast Cancer Research and Treatment (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
