
Abstract
Withaferin-A is a withanolide, predominantly present in Ashwagandha (Withania somnifera). It has been shown to possess anticancer activity in a variety of human cancer cells in vitro and in vivo. Molecular mechanism of such cytotoxicity has not yet been completely understood. Withaferin-A and Withanone were earlier shown to activate p53 tumor suppressor and oxidative stress pathways in cancer cells. 2,3-dihydro-3β-methoxy analogue of Withaferin-A (3βmWi-A) was shown to lack cytotoxicity and well tolerated at higher concentrations. It, on the other hand, protected normal cells against oxidative, chemical and UV stresses through induction of anti-stress and pro-survival signaling. We, in the present study, investigated the effect of Wi-A and 3βmWi-A on cell migration and metastasis signaling. Whereas Wi-A binds to vimentin and heterogeneous nuclear ribonucleoprotein K (hnRNP-K) with high efficacy and downregulates its effector proteins, MMPs and VEGF, involved in cancer cell metastasis, 3βmWi-A was ineffective. Consistently, Wi-A, and not 3βmWi-A, caused reduction in cytoskeleton proteins (Vimentin, N-Cadherin) and active protease (u-PA) that are essential for three key steps of cancer cell metastasis (EMT, increase in cell migration and invasion).
Similar content being viewed by others

Introduction
Cancer cell metastasis is a multistep process in which cancer cell acquires higher migration ability, invasive characteristics to permeate the basement membrane/extracellular matrix, access to lymphatic or vascular circulation towards developing secondary tumors at distant sites1. Metastatic malignancies have been shown to contribute to about 90% of cancer-related mortalities. Metastatic tumor cells, not only escape conventional radiotherapy, but also show a high level of resistance to a variety of chemotherapeutic combinations2. At molecular level, a cancer cell while acquiring metastatic ability, undergoes a drastic remodeling of cytoskeleton and membrane components3. Increase in vimentin (Type III intermediate filament protein), N-cadherin (transmembrane glycoprotein) and loss of E-cadherin have been characterized as key molecular alterations during Epithelial-to-Mesenchymal Transition (EMT), a primary step in cancer metastasis. A family of secretory proteases facilitates invasion of tumor cells through extracellular matrix and neighboring tissue compartments3,4. Therein, membrane-bound serine protease urokinase-type plasminogen activator (uPA) and matrix metalloproteinase (MMPs) proteases primarily degrade the extracellular matrix and facilitate extravasation of a tumor cells5. Process of angiogenesis mediated by secreted mitogens and vascular endothelial growth factor (VEGF), a key angiogenic factor, supports the formation of new blood vessels and promotes endothelial cell proliferation, cell migration, and survival6.
Withaferin A (Wi-A), is a potent bioactive withanolide that was first isolated from the roots of Ashwagandha (Withania somnifera), an Indian Ayurvedic medicinal herb. In initial investigations, Wi-A was shown to possess anticancer activity through diverse mechanisms including activation of caspase-3 and pERK, inhibition of JNK, Akt and IL6 pro-survival signaling7,8, instigation of oxidative stress and DNA damage response (DDR), leading to growth arrest or apoptosis in cancer cells in a dose-dependent manner9. Wi-A was shown to induce vimentin aggregation, inhibiting invasion and metastases of the breast cancer cells10. Bargagna-Mohan et al. showed that Wi-A alters vimentin assembly by covalent modification of its cysteine 328 residue11; later it was found to cause vimentin phosphorylation12. More recently, some studies showed that the treatment with Wi-A containing root extract inhibited metastases and EMT process13,14. We had earlier demonstrated that Wi-A and Withanone inhibit cancer cell migration and invasion by downregulating heterogeneous nuclear ribonucleoprotein-K (hnRNP-K), VEGF and MMPs proteins15.
2,3-dihydro-3β-methoxy withaferin-A (3βmWi-A) is a natural and structurally close Wi-A analogue, having substitution of β-methoxy group at position 3 of Wi-A ergostane ring16. Structural analogue of natural compounds, having chemical modification i.e. methylation, acetylation or hydroxylation at key moieties has been shown to affect their bioactivity, underlining structure-function association17. More precisely, alkali analogues of organic steroid compounds have been shown to attain improved potency18,19. Markedly, 10-methoxy derivative of Camptothecin, was shown to induce anti-angiogenic response20. Moreover, dimethoxy substitution to Brartemicin, a trehalose-based metastasis inhibitor, was shown to improve potency in murine colon carcinoma model21. Similarly, cis-resveratrol methylated analogs were shown to augment anti-tumor potency in metastatic mouse melanoma cells22. 2-methoxyestradiol, a natural estrogen analogue was characterized to be a potent anti-angiogenic compound23.
In light of these findings, we had earlier characterized the activities of Wi-A and its methoxy analogue 3βmWi-A. Unlike Wi-A, 3βmWi-A was found to be safe and well-tolerated at even higher concentration in vitro16. On contrary to Wi-A, 3βmWi-A was found to be inert and lacked cytotoxicity, and shown to promote survival of normal human cells exposed to various stress conditions24. Consistently and the fact that some natural compounds at nontoxic doses have been shown to possess anti-metastasis activity25,26, we investigated the effect of 3βmWi-A on cancer cell migration using bioinformatics and molecular assays in cultured cells. We report that whereas Wi-A binds to vimentin and hnRNP-K and modulate VEGF and MMPs signaling, 3βmWi-A lacked these activities.
Results
Wi-A, but not 3βmWi-A, inhibited cancer cell migration and invasion
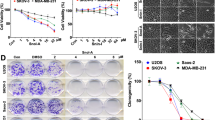
In order to investigate the effect of Wi-A and 3βmWi-A on cancer cell migration, we used their sub-toxic doses (0.3 and 0.6 μM)16,27. As shown in Fig. 1a, analyses of cell phenotype and viability showed no significant effect on U2OS (osteosarcoma) cells. In wound-healing assay, we found that whereas Wi-A inhibited U2OS cell migration at even the low dose (0.3 μM), 3βmWi-A did not cause any difference with respect to the control at both doses used (Fig. 1b). Quantitation of these data (Fig. 1b) showed dose-dependent and significant delay in migration of cells when treated with Wi-A, but not 3βmWi-A. To confirm that the anti-migration effect of Wi-A was not due to its cytotoxicity, time-lapse observations in control, Wi-A and 3βmWi-A (0.3 μM) treated GFP-labeled U2OS cells revealed that Wi-A, but not 3βmWi-A, delayed U2OS cell migration (data not shown); while no cell death was observed. We also confirmed the effect on three other metastatic cancer cell lines (MDA-MB-231, MCF-7 and HT-1080). As shown in Fig. 1c (photomicrographs), whereas sub-toxic doses of Wi-A caused significant delay in migration of all three cell types, 3βmWi-A remained inert. Quantitation of the data (Fig. 1c) showed Wi-A, not 3βmWi-A possesses significant anti-migratory activity. We also performed quantitative Matrigel invasion analyses that revealed a potent anti-invasion effect of Wi-A, but not of 3βmWi-A, both at 0.3 and 0.6 μM concentrations (Fig. 1d). Furthermore, a dose dependent effect (30 to 50% decrease) on invasiveness (both 0.3 and 0.6 μM) was observed in Wi-A treated U2OS cells. Conversely, 3βmWi-A did not exhibit anti-invasion activity. Therefore, these results suggested that Wi-A, and not 3βmWi-A, inhibited cancer cell migration and invasion (Fig. 1).

Wi-A, and not 3βmWi-A, inhibited cancer cell migration and invasion. (a) Representative phase contrast images showing cell morphology of control and Wi-A and 3βmWi-A (sub-toxic concentrations; 0.3 and 0.6 μM) treated cells. Quantitation of cell viability assay in U2OS control and treated cells at 48 h is shown. (b) Representative images from wound healing assay showing scratched area in control and Wi-A and 3βmWi-A treated U2OS cells at 0 h, 24 h and 48 h time points; while its quantitation showing percentage of gap filled at 48 h time point is shown in the lower panel. (c) Representative images from wound healing assay performed in metastatic MDA-MB-231, MCF7 and HT-1080 cell lines showing scratched area in control and 0.6 μM Wi-A and 3βmWi-A treated sets at 0 h, 24 h and 48 h time points; quantitation (lower panel) showing percentage covered area in Wi-A and 3βmWi-A treated cells (taking control as 100%) at 48 h. (d) Representative images from Matrigel invasion assay showing invaded crystal violet stained cells in control, Wi-A and 3βmWi-A treated cells. Quantitation on the right, showing percentage of invaded cells in control, Wi-A and 3βmWi-A treated cells.
Molecular docking of Wi-A and 3βmWi-A with Vimentin
In light of the information that Wi-A caused vimentin aggregation resulting in abrogation of invasion and metastases of the breast cancer cells10,11,12, we examined the interactions of these two molecules with vimentin by bioinformatics and molecular docking (MD) tools. The computationally modelled structure of vimentin dimer and tetramer was retrieved and vimentin monomer coordinates were extracted from dimer structure. MD simulation of vimentin modelled structure from residue 79–406 (monomer, dimer and tetramer) was carried out for 50 ns. The average representative structure of Vimentin was selected from stable simulation trajectories for each case. The RMSD of each structure was also analyzed to treat it as a control (Fig. S1a). The RMSD comparison of Vimentin monomer with its dimeric and tetrameric forms showed that Vimentin monomer is highly unstable whereas dimer and tetrameric forms have comparable and stable RMSD fluctuations. The interaction of Wi-A and 3βmWi-A was examined near Cys328 residue of Vimentin using molecular docking and simulation tools. Cys328 residue plays important role in reorganization of Vimentin network in response to oxidants as well as Vimentin polymerization by interacting with Zinc28. Interaction of any compounds at this critical residue may affect polymerization and assembly of Vimentin. Binding affinity and interactions formed by Wi-A and 3βmWi-A at the binding site of Vimentin monomer, dimer and tetramer are listed in Table 1. MM-GBSA was also used to find ligand binding affinity in docked complexes before and after MD simulations (Table 1).
Binding affinity of ligands reported by MM-GBSA were significant, hence, we proceeded with MD simulation of these complexes to investigate stability of interactions predicted at the binding site. Both molecules were found to be stable at the binding site of all forms of Vimentin, interacting with almost same residues but with slightly different orientations (Fig. 2a,b and Fig. S2a–d). Wi-A and 3βmWi-A did not show any interaction with Cys328, however, both interacted with its neighboring residues and could interfere with interaction of other entities with Cys328, thereby affecting polymerization and reorganization of Vimentin filaments. Next, we examined the effect of Wi-A and 3βmWi-A on stability of protein structure. RMSD computation of Vimentin monomer (Control) and in complex with Wi-A and 3βmWi-A showed that ligand binding around Cys328 increased the stability of Vimentin monomer structure, which was comparable to its dimeric and tetrameric forms (Fig. S1b). Of note, we found that Wi-A added more stability to Vimentin monomer (RMSD fluctuations in range of 20–30 Å) as compared to 3βmWi-A (that is 30–40Å). In line with this, the MM-GBSA calculated binding affinity of Wi-A (−49.61 Kcal/mol), was stronger than 3βmWi-A (−35.44 Kcal/mol). We also found that Wi-A formed both hydrophobic and hydrogen bond interactions, while 3βmWi-A interacted solely by hydrophobic interactions and did not form any hydrogen bonds (Fig. S2a,b). For Vimentin dimer and tetramer, RMSD fluctuations of Vimentin (control), Vimentin-Wi-A complex and Vimentin-3βmWi-A complex were comparable (Fig. S1c). These data suggested that Wi-A and 3βmWi-A may not affect stability of Vimentin dimer and tetramer. In case of Vimentin dimer, 3βmWi-A showed stronger binding efficiency by laterally interacting with both chains of dimer (binding energy of −61 Kcal/mol). On the other hand, Wi-A showed longitudinal interactions with chain A only (binding energy of −37.59 Kcal/mol) (Fig. S2c,d). In addition, both Wi-A and 3βmWi-A formed hydrophobic interaction with Gln322, a conserved residue found in the highly conserved domain protein family of intermediate filament (Fig. S3)29. Similarly, in case of Vimentin tetramer both Wi-A and 3βmWi-A showed binding at the same site forming interactions with almost same residues with slightly different binding affinity of −93.51 and −78.48 Kcal/mol, respectively (Fig. 2a,b). Both Wi-A and 3βmWi-A interacted with conserved residue Gln322. However, we found that Wi-A caused local disruption of helical structure at the binding site while no such effect was observed in case of 3βmWi-A. Such configurations are predicted to affect interaction of Vimentin with metal ions (Zn) for formation of Vimentin octamer28. These data suggested that both Wi-A and 3βmWi-A can interact with Vimentin monomeric, dimeric and tetrameric forms. Wi-A, however, showed high binding affinity for and also caused disruption of secondary structure at the binding site of vimentin tetramer. Hence, it was predicted that Wi-A, but not 3βmWi-A, targets Vimentin causing anti-migration and anti-metastatic effects.

Molecular docking of Wi-A and 3βmWi-A with Vimentin. Molecular interactions of vimentin tetramer at the binding site around Cys328 with Wi-A (a) and 3βmWi-A (b).
We next performed molecular analyses to confirm the above results. As shown in Fig. 3a, we found significant decrease in Vimentin, a key intermediate filament protein involved in cell migration, in Wi-A, but not 3βmWi-A, treated cells. We, next, analyzed the localization and assembly of Vimentin in control and treated cells. Whereas Wi-A (0.6 μM) treated cells showed distinct Vimentin aggregation (Fig. 3b), there was no difference in response to 3βmWi-A treatment. The results were confirmed with high resolution confocal microscopy (Fig. 3b). Dose dependent effect of Wi-A and 3βmWi-A, and analysis of Vimentin expression by immunostaining further revealed its significant reduction in Wi-A treated cells, while its levels remain unchanged in response to 3βmWi-A treatment (Fig. 3b,c). Based on these data and Figs. 1 and 2, it was confirmed that whereas Wi-A targeted Vimentin protein, 3βmWi-A lacks such activity. The data was consistent to an earlier report that showed Vimentin as a target of Wi-A10. In order to get further insights to the differential bio-activities of Wi-A and 3βmWi-A, we examined their effect on the expression of N-cadherin and E-cadherin, two key mesenchymal and epithelial cell markers. As shown in Fig. 3d, Wi-A, but not 3βmWi-A, caused decrease in N-cadherin in dose-dependent manner (Fig. 3d). In contrast, there was no change in the level of E-cadherin as validated by immunoblotting (Fig. 3e) and immunostaining (Fig. 3f), respectively. These data suggested that Wi-A has potent anti-migratory activities, while 3βmWi-A lacks such effect.

Wi-A, and not 3βmWi-A, affected Vimentin and N-Cadherin. (a) Immunoblotting (upper panel) and its quantitation (lower panel) showing Vimentin expression levels in control, 0.3 μM, and 0.6 μM Wi-A and 3βmWi-A (cropped blot omitting their 1.5, and 3 μM toxic doses) treated U2OA cancer cells. (b) Immunostaining showing vimentin expression and distribution in treated cells. Inset image (lower panel) (marked by white arrow), showing vimentin aggregates in 0.6 μM Wi-A cells, though no similar aggregates were observed in control and 3βmWi-A treated cells. (c,d) Vimentin and N-cadherin immunostaining showing their expression levels in control and 0.3, 0.6 μM Wi-A and 3βmWi-A treated cells; their quantitation is shown on the right. (e) Immunoblot showing unaltered E-cadherin expression in Wi-A and 3βmWi-A treated cells (cropped blot omitting their 1.5, and 3 μM toxic doses); quantitation of its expression is shown in the lower panel. (f) Immunostaining showing unaltered E-cadherin expression in treated cells; quantitation shown at the bottom.
Interactions of Wi-A and 3βmWi-A with the KH3 domain of hnRNP-K and functional consequences
In light of the above findings and our earlier report that Wi-A targets hnRNP-K protein15, we next examined the effect of 3βmWi-A on hnRNP-K activity. Molecular docking and simulation analyses using KH3 domain of hnRNP-K that has been shown to bind to ssDNA revealed strong binding strength of Wi-A (docking score Wi-A −4.54 Kcal/mol) as compared to 3βmWi-A (docking score 3βmWi-A −3.17 kcal/mol) (Fig. 4a,b,e). Although the two withanolides shared similar configuration and surrounding residues, analysis of their superimposed interactions with hnRNP-K 3D-structure demonstrated that β-methoxy group of 3βmWi-A at C3 position caused steric hindrance and hence decreased its binding strength as compared to Wi-A (Fig. 4c,d). As shown in Fig. 4a,b,e, the superimposed images on the binding of Wi-A and 3βmWi-A with the ssDNA-binding interface revealed weaker docking capability of 3βmWi-A as compared to Wi-A. Following these results, we next examined the expression of hnRNP-K in control and treated cells. As shown in Fig. 5a, immunoblotting revealed dose-dependent decrease in its protein levels in Wi-A, but not 3βmWi-A, treated cells. Furthermore, immunostaining analysis also revealed a decrease in hnRNP-K expression levels in Wi-A treated cells (Fig. 5b). Of note, no change in hnRNP-K transcript levels were observed in Wi-A or 3βmWi-A treated cells (Fig. 5c, below).

Interactions of Wi-A and 3βmWi-A with the KH3 domain of hnRNP-K. (a) Molecular modeling showing binding of Wi-A (green) at the DNA binding interface of hnRNP-K. (b) Molecular binding of 3βmWi-A (yellow) at the DNA binding interface of hnRNP-K. (c) Table showing details of docking score and predicted molecular interactions of hnRNP-K with Wi-A & 3βmWi-A. (d) Superimposed docking conformations of Wi-A (green) and 3βmWi-A (yellow), bound at KH3 domain of hnRNP-K. (e) Superimposed Wi-A (green), 3βmWi-A (yellow) and ssDNA (red) at the DNA binding KH3 domain of hnRNP-K.

Wi-A, and not 3βmWi-A, inhibited hnRNP-K at protein levels. (a) Immunoblot showing hnRNP-K expression in control, 0.3 μM, and 0.6 μM Wi-A and 3βmWi-A (cropped blot omitting their 1.5, and 3 μM toxic doses) treated U2OS cancer cells; quantitation of its expression is shown in the lower panel. (b) Immunostaining showing hnRNP-K expression in the treated cells; quantitation of its expression is shown at the bottom. (c) RT-PCR showing hnRNP-K transcript in the treated cells; quantitation of its expression is shown in the lower panel.
Wi-A, but not 3βmWi-A, inhibits hnRNP-K-VEGF signaling
In light of the fact that hnRNP-K is a key regulator of VEGF and promotes its mRNA stability and translation30,31, we next prompted to examine VEGF level in Wi-A and 3βmWi-A treated cells. As expected, Wi-A, but not 3βmWi-A, treated cells showed decrease in VEGF in immunostaining analyses (Fig. 6a). It was further confirmed by immunoblotting analyses that endorsed decrease in the VEGF protein in Wi-A treated cells only (Fig. 6b). Of note, 3βmWi-A treated cells, showed higher level of VEGF protein (Fig. 6a,b). VEGF transcript, on the other hand, showed no difference either in Wi-A or 3βmWi-A treated cells (Fig. 6c) suggesting that Wi-A may essentially affects VEGF at the protein level. Consistent to these results, we next examined the level of secreted VEGF in treated cells by multiplex immunoarray and found that whereas Wi-A caused decrease in secreted VEGF levels, it was increased by more than two-fold in 3βmWi-A treated cells (Fig. 7a). Interestingly, IL-10, IL-12, IL-13, key interleukin factors regulating VEGF, levels also showed decrease in response to Wi-A treatment and increase in 3βmWi-A treatment (Fig. 7a). In order to examine the protein targets of Wi-A relevant to cancer cell invasion and metastases effects, we next performed MMP Antibody array. Untreated U2OS cells were used as negative control. As shown in Fig. 7b, Wi-A treated cells showed remarkable decrease in MMP1, MMP2, MMP8, MMP9, MMP10 and MMP13 expression (Fig. 7b). On the other hand, 3βmWi-A treated cells exhibited a slight and insignificant change in their levels, when normalized with untreated control (Fig. 7b). Expression analyses of VEGF downstream effectors viz. u-PA32 and MMP233 that play vital role in cancer cell migration, also exhibited their reduced levels in Wi-A, but not 3βmWi-A treated cells (Fig. 7c). The data suggested that Wi-A, consistent to the downregulation of hnRNPK-VEGF signaling, suppressed expression of several key MMPs/proteases involved in cell invasion and metastases, wherein 3βmWi-A remained largely ineffective. Also, we examined VEGF downstream targets phospho-p38MAPK and hsp27, two key VEGF markers promoting cancer cell survival during metastases/angiogenesis. As shown in Fig. 7d, a dose-dependent decrease in phospho-p38MAPK and hsp27 proteins was observed in Wi-A, but not in 3βmWi-A treated cells.

Wi-A, and not 3βmWi-A, reduced the level of VEGF expression. (a) Immunostaining showing VEGF expression in control and 0.3, 0.6 μM Wi-A and 3βmWi-A treated U2OS cancer cells; quantitation of its expression is shown in the lower panel. (b) Immunoblot showing VEGF expression in Wi-A and 3βmWi-A treated cells (cropped blot omitting their 1.5, and 3 μM toxic doses); quantitation of its expression is shown below. (c) RT-PCR showing VEGF transcript in the treated cells; quantitation of its expression is shown in the lower panel.

Wi-A, and not 3βmWi-A, inhibits hnRNP-K-VEGF signaling. (a) Multiplex immunoarray showing quantitative levels of VEGF and IL-10, 12, 13 pro-metastases secretory proteins in Wi-A and 3βmWi-A treated cells; expression values were normalized with untreated control condition media and represented as fold change. (b) MMP Antibody Array showing quantitative expression levels of MMP1, 2, 8, 9, 10 and 13 proteins in the condition media of the Wi-A and 3βmWi-A treated U2OS cells, expression values normalized with untreated control and represented as fold change. (c) Immunoblot showing expressions of uPA and MMP2 in Wi-A and 3βmWi-A treated cells (cropped blot omitting their 1.5, and 3 μM toxic doses); quantitation of their expressions is shown in the lower panel. (d) Immunoblot showing expressions of phospho-p38MAPK and hsp27 in Wi-A and 3βmWi-A treated cells (cropped blot omitting their 1.5, and 3 μM toxic doses); quantitation of their expressions is shown at the bottom.
Discussion
Wi-A and 3βmWi-A are natural co-occurring withanolides, isolated from Ashwagandha. 3βmWi-A, with the substitution of a β-methoxy group at Wi-A parental ring is suggested to be a natural Wi-A analogue. Wi-A is potent bioactive compound, and has been shown to inhibit metastases and EMT process in vitro and in vivo models essentially by promoting Vimentin aggregation12,13,14,15,16 and regulating hnRNP-K function17. With evident anti-metastatic potency of Wi-A, and the concept that the alkali analogues retain augmented bioactivities18,19; we investigated the activities of 3βmWi-A on cancer cell migration at cellular and molecular levels. Taking the sub-toxic concentrations that showed no effect on cell viability, we found Wi-A, but not 3βmWi-A, inhibited cancer cell migration and invasion. By molecular docking and dynamic simulation studies, Wi-A was predicted to possess greater binding affinity to Vimentin and tetramer than 3βmWi-A. It also caused local disruption of helical structure at Vimentin tetramer binding site. Consistently, Wi-A, but not 3βmWi-A, led to Vimentin aggregation. Wi-A treated cells showed decrease in Vimentin, N-cadherin and E-cadherin proteins, signifying compromised cell migration and invasion34. Wi-A was earlier shown to target hnRNP-K, a key regulator of cell migration and angiogenesis15,30. Inputs from in silico analysis confirmed a stronger binding of Wi-A, over 3βmWi-A, at the hnRNPK’s KH3 domain i.e. ssDNA binding domain. It could lead to instability and decrease in hnRNP-K and its downstream effectors including VEGF and MMPs leading to compromised cell migration15,32,35,36. Reduced levels of MMPs, uPA proteases and VEGF targets viz. phospho-p38MAPK37, heat shock protein 27 (HSP27) that promote cancer cell migration37,38,39, were indeed found in Wi-A, but not 3βmWi-A, treated cells. Taken together, we demonstrate that 3βmWi-A lacked cytotoxicity to cancer cells and showed no effect on cell migration, invasion and angiogenic characteristics. Wi-A was found to affect the Vimentin assembly, yielding its decrease in treated cells; 3βmWi-A was ineffective. Wi-A, as compared to 3βmWi-A, also showed stronger binding to hnRNP-K. Additionally, it caused decrease in other key regulators of cell migration, invasion and metastasis, viz, MMPs and VEGF, u-PA, Hsp27 and p38MAPK. Taken together, we found that in contrast to Wi-A and the anticipation that alkali analogues may exhibit stronger potency, 3βmWi-A lacked anti-metastatic activity. Hence, structural-function predictions of natural compounds warrant careful experimental validation before their recruitment in disease preventive or therapeutic avenues.
Methods
Cell culture
Osteosarcoma (U2OS; U2OS-GFP), fibrosarcoma (HT-1080) and breast adenocarcinoma (MBA-MB-231, MCF-7) cell lines were procured from Japanese Collection of Research Bioresources Cell Bank (JCRB), Tokyo, and cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Wako, Japan) supplemented with 10% Fetal Calf Serum (FCS) and 1% antibiotics in a humidified incubator containing 5% CO2 at 37 °C as described earlier40.
Cell viability assay
5 × 103 cells were seeded in a 96-well plate and allowed to adhere in next 24 h. On the following day, cells were treated with 0.3, 0.6 μM of Wi-A and 3βmWi-A for next 48 h. Tetrazolium dye 3-(4,5-dimethylthiazol-2-yl)−2,5-diphenyltetrazolium bromide (MTT reagent, Invitrogen, Life technologies), was used to determined viability of both control and Wi-A, 3βmWi-A treated cells, as described earlier in details16.
Immunoblotting
Cells pellets were lysed in RIPA buffer (Sigma-Aldrich). Supernatant containing 20 µg of proteins was resolved in SDS-polyacrylamide gel and electroblotted onto PVDF membranes (Millipore, Billerica, MA) using a semidry transfer unit (ATTO, Tokyo, Japan). Immunoblotting was performed with anti-VEGF, MMP-2, Vimentin (Santa Cruz), N-Cadherin, uPA & hnRNP-K (Abcam), phospho-p38MAPK, E-Cadherin (Cell Signaling), HSP27 (StressMarq Biosciences Inc.) and β-actin (Abcam, Cambridge) antibodies. The membranes probed with the first antibodies were excessively washed with TBS-T (Tris-buffered saline-Tween 20) and incubated with secondary horseradish peroxidase (HRP)-conjugated goat anti-mouse or anti-rabbit (Santa Cruz) antibodies. Protein bands were detected using ECL prime substrate (GE Healthcare, CA). Densitometric quantitation of three independent immunoblotting experiments was performed with the Image J software (NIH, Bethesda, MD). Expression level of each of the proteins in control and treated cells was calculated with respect to the β-actin (loading control). All experiments were performed in triplicate.
Immunofluorescence staining
Cells were cultured as described above. 4 × 104 cells /well were seeded on glass coverslips placed in a 12-well plate for 24 h. Cells were treated with indicated doses of either Wi-A or 3βmWi-A for 24 h and then fixed with pre-chilled methanol on ice for 10 min. PBS-Triton-X-100 (0.2%) for 10 min were used to permeablized the fixed cells followed by blocking with 2% BSA (10 min). The coverslips were probed with antibodies against VEGF, hnRNP-K, Vimentin, N-Cadherin and E-Cadherin at room temperature (1 h) or 4 °C (overnight). Cells were then probed with Alexa Fluor-conjugated secondary antibodies (Molecular Probes, Invitrogen), and counterstained with Hoechst 33258 (Thermo Fisher Scientific). The stained cells were viewed under Zeiss Axioplan 2 microscope and images were captured using a Zeiss AxioCam HRc camera. Acquired images were quantified using ImageJ software, while fluorescence intensity values were normalized with respective controls and represented in percentage values. To examine the distribution and aggregation of Vimentin in Wi-A or 3βmWi-A treated cells, images were acquired under confocal laser scanning microscope (ZEISS LSM 700) and analyzed by IMARIS® software (Bitplane, Switzerland).
Wound healing assay
Motility of cells was examined using the Wound-healing assay. 2.5 × 105 cells/well were seeded in 6-well plate and allowed to attach to the substratum for next 24 h. Monolayers of cells were wounded on the following day by uniformly scratching the surface with a scrapper tip (20 gauge), followed by washing with PBS and then fresh medium was added. Cells were allowed to proliferate and migrate into the wound for, at least, 24 or 48 h. Movement of the control and treated cells in the scratched area were serially monitored under a phase contrast microscope with a 10 X phase objective (Nikon). Time-lapse live imaging was performed using a microscope (Carl Zeiss MicroImaging, ApoTome) equipped with a Plan-Apochromat 10x/0.45 DICII objective, motorized scanning table and a stage incubator at 37 °C with CO2. Images were captured with an AxioCam MR Rev 3 Monochromatic Digital Camera using the AxioVision rel 4.8 software for microscope control and data acquisition. Time-point captured images were processed with image J software adjusting contrast and color threshold options for representation. Percentage covered area was calculated using % area option and crosschecked with analyze particle options using normalized processed images (6–10 randomly captured). The assay was repeated in triplicate in parallel to the Cell Viability Assay.
Matrigel cell invasion assay
4 × 103 cells were seeded into the upper chamber, coated on the surface with 1/10 dilution of Matrigel (BD BioSciences, Franklin Lakes, NJ), and allowed to migrate to the lower chamber as described in details earlier41. Migrated cells were fixed and stained with crystal violet and counted under phase contrast microscopy.
Reverse-transcription PCR (RT-PCR)
RNA was extracted from control, Wi-A or 3βmWi-A treated cells with Qiagen RNeasy kit. cDNA was synthesized from 2 μg of RNA using the ThermoScript Reverse Transcriptase (ThermoFisher Scientific) following the manufacturer’s protocol. cDNA was subjected to PCR amplification along with gene specific sense and antisense primer set (as follows) using TaKaRa Ex Taq® DNA polymerase, steps consisting of an initial 10 min denaturation step at 95 °C followed by 34 cycles at 95 °C for 45 s, 56 °C for 1 min and 72 °C for 45 s, with final annealing step at 72 °C for 10 min. PCR amplifications were performed using specific primers for (i) hnRNP-K: Sense: 5′-ATGAAATTCACCCCCTTTCC-3′ & Antisense: 5′-CCCTAGGCTGTGCTCACTTC-3′, (ii) VEGF- Sense: 5′-CTGCCCTCAACAAGATGTTTTG-3′ & Antisense: 5′-CTATCTGAGCAGCGCTCATGG-3′, (iii) GAPDH: Sense: 55-ACCTGACCTGCCGTCTAGAA-3′ & Antisense: 5′-TCCACCACCCTGTTGCTGTA-3′. The PCR products were then run on a 1% agarose gel and stained with ethidium bromide for visualization.
Molecular interaction and docking analysis
Molecular docking and simulations were performed using modules of Schrodinger 2018-2 suite. The structure of proteins and ligands (Wi-A and 3βmWi-A) were prepared using PrepWizard and LigPrep module respectively42. Prepared structures were docked with ligands using Glide extra precision (XP) algorithm42,43. The molecular dynamic (MD) simulation of docked complexes was performed to check the stability of ligand interactions at the binding site using Desmond module42. All complexes were simulated in a SPC solvated periodic box with 10 Å spacing in Optimized Potential for Liquid Simulations 3 (OPLS3) force field. The solvated system was neutralized, minimized for up to 2000 iterations, heated to 300 K, equilibrated and simulated for a time period ranging between 40 to 50 ns.
-
i.
Molecular interaction study of Wi-A and 3βmWi-A with hnRNP-K KH3 domain. Tertiary structure of hnRNP-K KH3 domain with complex to ssDNA was obtained from Protein Data Bank (PDBID: 1ZZI). DNA binding interface of KH3 domain was used to generate grid, so that Wi-A and 3βmWi-A could be screened for their docking potentials at this site43. Prepared protein and ligands were then docked using XP docking protocol of Glide module. During docking, hydrogens of aromatic rings were also allowed to form hydrogen bonds.
-
ii.
Molecular interaction study of Wi-A and 3βmWi-A with monomer, dimer and tetrameric forms of Vimentin. Since X-ray crystallography structure of full Vimentin is not available so far, we used computational model of Vimentin dimer and tetramer structure generated by Qin et al., 200944. In order to investigate the effect of Wi-A and 3βmWi-A, first MD simulation of structure of Vimentin monomer, dimer and tetramer was carried out for 50 ns each. Simulated and stabilized Vimentin monomer, dimer and tetramer molecules were docked with Wi-A and 3βmWi-A around Cys328 residue using Glide extra precision (XP) algorithm.
The stability of inhibitors at the binding site was examined by performing molecular dynamic (MD) simulations of docked complexes. Vimentin monomer-ligand complexes and Vimentin dimer-ligand complexes were simulated for 50 ns each, while Vimentin tetramer-ligand complexes were simulated for 40 ns each. Prime MM-GBSA module of Schrodinger was used to calculate binding energy of protein-ligand complex42. Ligplus program was used to study protein-ligand interactions45. Root mean square deviation (RMSD), hydrogen bonds analysis and conformational changes over the simulation trajectories of protein-ligand complexes were monitored using VMD version 1.9.446. Images for publication were generated using Pymol molecular graphics system47.
Human MMP Antibody Array. To examine expression levels of Matrix metalloproteinase family proteins, Human MMP Antibody Array - membrane (#ab134004) were used to determine the total secreted MMP protein quantity in condition media of U2OS cells treated with Wi-A and 3βmWi-A (0.6 μM) following the manufacturer’s protocol.
Multiplex immunoassay
Bio-Plex Suspension Array System (Bio-Rad, Hercules, CA) was used for multiplex immunoassay profiling. Conditioned medium was collected from control and Wi-A or its methoxy analogue i.e. 3βmWi-A (0.6 μM) treated cells. Concentrations of the cytokines and growth factors in the medium were measured with respect to the standards prepared for each of the factors. Analysis of data was performed with the Bio-Plex Manager software (Bio-Rad), normalized to the control and represented as fold change over control.
Statistical analysis
All the experiments were carried out in triplicate. Data values were expressed as mean ± SEM of three individual experiment sets. Statistical analyses were carried out using Student’s t-test or nonparametric Mann-Whitney U-test; whichever was applicable. Statistical significance was defined as p-value ≤ 0.05. The p value represents * < 0.05, **< 0.01, ***< 0.001.
References
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Eccles, S. A. & Welch, D. R. Metastasis: recent discoveries and novel treatment strategies. Lancet 369, 1742–1757 (2007).
Alizadeh, A. M., Shiri, S. & Farsinejad, S. Metastasis review: from bench to bedside. Tumour Biol. 35, 8483–8523 (2014).
Klein, C. A. Selection and adaptation during metastatic cancer progression. Nature 501, 365–372 (2013).
Wang, Z. et al. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 69, 2400–2407 (2009).
Saharinen, P., Eklund, L., Pulkki, K., Bono, P. & Alitalo, K. VEGF and angiopoietin signaling in tumor angiogenesis and metastasis. Trends Mol. Med. 17, 347–362 (2011).
Lee, I. C. & Choi, B. Y. Withaferin-A–A Natural Anticancer Agent with Pleitropic Mechanisms of Action. Intl. J. Mol. Sci. 17, 290 (2016).
Oh, J. H. et al. Induction of apoptosis by withaferin A in human leukemia U937 cells through down-regulation of Akt phosphorylation. Apoptosis 13, 1494–1504 (2008).
Chang, H. W. et al. Withaferin A Induces Oxidative Stress-Mediated Apoptosis and DNA Damage in Oral Cancer Cells. Front. Physiol. 8, 634 (2017).
Thaiparambil, J. T. et al. Withaferin A inhibits breast cancer invasion and metastasis at sub-cytotoxic doses by inducing vimentin disassembly and serine 56 phosphorylation. Intl. J. Cancer 129, 2744–2755 (2011).
Bargagna-Mohan, P. et al. The tumor inhibitor and antiangiogenic agent withaferin A targets the intermediate filament protein vimentin. Chem. Biol. 14, 623–634 (2007).
Bargagna-Mohan, P. et al. Withaferin A effectively targets soluble vimentin in the glaucoma filtration surgical model of fibrosis. PLoS One 8, e63881 (2013).
Lee, D. H. et al. Withaferin A inhibits matrix metalloproteinase-9 activity by suppressing the Akt signaling pathway. Oncol. Rep. 30, 933–938 (2013).
Yang, Z. et al. Withania somnifera root extract inhibits mammary cancer metastasis and epithelial to mesenchymal transition. PLoS One 8, e75069 (2013).
Gao, R. et al. Withanone-rich combination of Ashwagandha withanolides restricts metastasis and angiogenesis through hnRNP-K. Mol. Cancer Therap. 13, 2930–2940 (2014).
Chaudhary, A. et al. 2,3-Dihydro-3beta-methoxy Withaferin-A Protects Normal Cells against Stress: Molecular Evidence of Its Potent Cytoprotective Activity. J. Nat. Prod. 80, 2756–2760 (2017).
Havsteen, B. H. The biochemistry and medical significance of the flavonoids. Pharmacol. Ther. 96, 67–202 (2002).
Sy-Cordero, A. A. et al. Enhanced bioactivity of silybin B methylation products. Bioorganic Med. Chem. 21, 742–747 (2013).
Walle, T. Methylation of dietary flavones increases their metabolic stability and chemopreventive effects. Intl. J. Mol. Sci. 10, 5002–5019 (2009).
Han, M. et al. A novel camptothecin derivative incorporated in nano-carrier induced distinguished improvement in solubility, stability and anti-tumor activity both in vitro and in vivo. Pharmaceut. Res. 26, 926–935 (2009).
Jiang, Y. L. et al. Synthesis and evaluation of trehalose-based compounds as anti-invasive agents. Bioorg. Med. Chem. Lett. 21, 1089–1091 (2011).
Morris, V. L. et al. Anti-tumor properties of cis-resveratrol methylated analogs in metastatic mouse melanoma cells. Mol. Cellular Biochem. 402, 83–91 (2015).
Peyrat, J. F., Brion, J. D. & Alami, M. Synthetic 2-methoxyestradiol derivatives: structure-activity relationships. Curr. Med. Chem. 19, 4142–4156 (2012).
Huang C et al. 3beta-Methoxy Derivation of Withaferin-a Attenuates its Anticancer Potency: Bioinformatics and Molecular Evidences. Med. Aromatic Plants 4 (2015).
Chaotham, C., Pongrakhananon, V., Sritularak, B. & Chanvorachote, P. A Bibenzyl from Dendrobium ellipsophyllum inhibits epithelial-to-mesenchymal transition and sensitizes lung cancer cells to anoikis. Anticancer Res. 34, 1931–1938 (2014).
Ham, S. L. et al. Phytochemicals potently inhibit migration of metastatic breast cancer cells. Integr. Biol. (Camb) 7, 792–800 (2015).
Vaishnavi, K. et al. Differential activities of the two closely related withanolides, Withaferin A and Withanone: bioinformatics and experimental evidences. PLoS One 7, e44419 (2012).
Pérez-Sala, D. et al. Vimentin filament organization and stress sensing depend on its single cysteine residue and zinc binding. Nature Commun. 6, 7287 (2015).
Marchler-Bauer, A. et al. CDD: NCBI’s conserved domain database. Nuc. Acids Res. 43, D222–D226 (2014).
Gao, R. et al. Heterogeneous nuclear ribonucleoprotein K (hnRNP-K) promotes tumor metastasis by induction of genes involved in extracellular matrix, cell movement, and angiogenesis. J. Biol. Chem. 288, 15046–15056 (2013).
Feliers, D., Lee, M. J., Ghosh-Choudhury, G., Bomsztyk, K. & Kasinath, B. S. Heterogeneous nuclear ribonucleoprotein K contributes to angiotensin II stimulation of vascular endothelial growth factor mRNA translation. Am. J. Physiol. Renal. Physiol. 293, F607–615 (2007).
Breuss, J. M. & Uhrin, P. VEGF-initiated angiogenesis and the uPA/uPAR system. Cell Adh. Migr. 6, 535–615 (2012).
Rodrigues, M. et al. VEGF secreted by hypoxic Muller cells induces MMP-2 expression and activity in endothelial cells to promote retinal neovascularization in proliferative diabetic retinopathy. Diabetes 62, 3863–3873 (2013).
Tsai, J. H. & Yang, J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes & Develop. 27, 2192–2206 (2013).
Sataranatarajan, K., Lee, M. J., Mariappan, M. M. & Feliers, D. PKCdelta regulates the stimulation of vascular endothelial factor mRNA translation by angiotensin II through hnRNP K. Cellular Signalling 20, 969–977 (2008).
Deryugina, E. I. & Quigley, J. P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 25, 9–34 (2006).
Rousseau, S., Houle, F., Landry, J. & Huot, J. p38 MAP kinase activation by vascular endothelial growth factor mediates actin reorganization and cell migration in human endothelial cells. Oncogene 15, 2169–2177 (1997).
Sawada, J., Li, F. & Komatsu, M. R-Ras Inhibits VEGF-Induced p38MAPK Activation and HSP27 Phosphorylation in Endothelial Cells. J. Vasc. Res. 52, 347–359 (2015).
Yoshizuka, N. et al. A novel function of p38-regulated/activated kinase in endothelial cell migration and tumor angiogenesis. Mol. Cellular Biol. 32, 606–618 (2012).
Kalra, R. S. et al. CARF enrichment promotes epithelial-mesenchymal transition via Wnt/beta-catenin signaling: its clinical relevance and potential as a therapeutic target. Oncogenesis 7, 39 (2018).
Kalra, R. S. et al. CARF (Collaborator of ARF) overexpression in p53-deficient cells promotes carcinogenesis. Mol. Oncol. 9, 1877–1889 (2015).
Protein Preparation Wizard; Desmond Molecular Dynamics System, D. E. Shaw Research, Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2018. v. 2 (Schrödinger Release 2018-2).
Friesner, R. A. et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49, 6177–6196 (2006).
Qin, Z., Kreplak, L. & Buehler, M. J. Hierarchical structure controls nanomechanical properties of vimentin intermediate filaments. PLoS One 4, e7294 (2009).
Wallace, A. C., Laskowski, R. A. & Thornton, J. M. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng., Design Selection 8, 127–134 (1995).
Humphrey, W., Dalke, A. & Schulten, K. VMD - Visual Molecular Dynamics. J. Molec. Graphics 14, 33–38 (1996).
DeLano, W. L. The PyMOL molecular graphics system, http://www/pymol.org (2010).
Acknowledgements
The work was supported by funds from the Department of Biotechnology (DBT), Govt. of India, India, and the National Institute of Advanced Industrial Science & Technology (AIST), Tsukuba, Japan. The Computations were performed at the Bioinformatics Centre at IIT Delhi, India.
Author information
Authors and Affiliations
Contributions
R.S.K., V.M., D.S. and R.W. designed the study. A.C. and R.S.K. performed experiments.V.M. and S.P.K. performed computational analyses. R.S.K., V.M., D.S., S.C.K. and R.W. wrote the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chaudhary, A., Kalra, R.S., Malik, V. et al. 2, 3-Dihydro-3β-methoxy Withaferin-A Lacks Anti-Metastasis Potency: Bioinformatics and Experimental Evidences. Sci Rep 9, 17344 (2019). https://doi.org/10.1038/s41598-019-53568-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53568-6
This article is cited by
-
Withaferin A mitigates metastatic traits in human oral squamous cell carcinoma caused by aberrant claudin-1 expression
Cell Biology and Toxicology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
