
Abstract
The anaerobic gut pathogen, Clostridioides difficile, forms adherent biofilms that may play an important role in recurrent C. difficile infections. The mechanisms underlying C. difficile community formation and inter-bacterial interactions are nevertheless poorly understood. C. difficile produces AI-2, a quorum sensing molecule that modulates biofilm formation across many bacterial species. We found that a strain defective in LuxS, the enzyme that mediates AI-2 production, is defective in biofilm development in vitro. Transcriptomic analyses of biofilms formed by wild type (WT) and luxS mutant (luxS) strains revealed a downregulation of prophage loci in the luxS mutant biofilms compared to the WT. Detection of phages and eDNA within biofilms may suggest that DNA release by phage-mediated cell lysis contributes to C. difficile biofilm formation. In order to understand if LuxS mediates C. difficile crosstalk with other gut species, C. difficile interactions with a common gut bacterium, Bacteroides fragilis, were studied. We demonstrate that C. difficile growth is significantly reduced when co-cultured with B. fragilis in mixed biofilms. Interestingly, the absence of C. difficile LuxS alleviates the B. fragilis-mediated growth inhibition. Dual species RNA-sequencing analyses from single and mixed biofilms revealed differential modulation of distinct metabolic pathways for C. difficile WT, luxS and B. fragilis upon co-culture, indicating that AI-2 may be involved in induction of selective metabolic responses in B. fragilis. Overall, our data suggest that C. difficile LuxS/AI-2 utilises different mechanisms to mediate formation of single and mixed species communities.
Similar content being viewed by others


Introduction
Clostridiodes difficile (Clostridium difficile), an anaerobic, opportunistic pathogen, is the causative agent of C. difficile infection (CDI), a debilitating condition with symptoms ranging from mild diarrhoea to severe pseudomembranous colitis. ~453,000 cases of CDI were reported in the United States in 20111 and there have been increasing reports of CDI from different parts of the world2,3,4. Treatment of CDI is complicated by the fact that 20–36% of cases experience recurrence, relapsing after completion of initial treatment5. CDI is primarily a hospital-acquired infection with the elderly being at highest risk6 and has been associated with the disruption of the gut microbiota as a result of the use of broad-spectrum antibiotics. However, more recently, there has been a reported increase in community-acquired cases where patients do not have the typical risk factors such as antibiotic exposure or recent hospitalisation7.
Colonisation of C. difficile and development of CDI is influenced by composition of the native gut microbiota. Broadly, Bacteroides, Prevotella, Bifidobacterium, Enterococcaceae and Leuconostocaceae spp. correlate negatively8,9,10, and Lactobacilli, Aerococcaceae, Enterobacteriaceae, and Clostridium correlate positively to C. difficile colonisation and disease8,10,11,12,13. While mechanisms underlying colonisation resistance are not entirely clear, some pathways have been described recently. Secondary bile acids produced by bacteria like Clostridium scindens can inhibit C. difficile growth, while other bile acids such as chenodeoxycholate can inhibit spore germination14,15,16. Studies have shown that the ability of C. difficile to utilise metabolites produced by the gut microbiota or mucosal sugars such as sialic acid promote C. difficile expansion in the gut17,18. However, gaps still remain in our understanding of C. difficile interactions with members of the gut microbiota.
Research into CDI has primarily focused on the action of two large toxins19,20 that cause tissue damage, neutrophil recruitment and a severe inflammatory response21. More recently, a number of factors have been shown to influence adhesion of C. difficile to host cells and early colonisation, including cell wall proteins, adhesins and flagella22,23,24,25,26. C. difficile also produces biofilms that confer increased resistance to antibiotics27,28,29 and have recently shown to be associated with C. difficile infection in vivo, in close association with other commensal gut species30.
Formation of adherent communities within the gut requires communication between bacteria. For many species, quorum sensing (QS) is important for the construction and/or dispersal of biofilm communities31, with bacteria utilising diverse QS systems31,32. Many bacteria possess the metabolic enzyme LuxS, which is involved in the detoxification of S-adenoslylhomocysteine during the activated methyl cycle. Whilst catalysing the reaction of S-ribosylhomocysteine to homocysteine, LuxS produces the bi-product 4,5-dihydroxy-2,3-pentanedione (DPD). DPD is an unstable compound that spontaneously cyclises into several different forms. These ligands are collectively known as autoinducer-2 (AI-2), a group of potent, cross-species QS signalling molecules33. In many bacteria, including C. difficile, AI-2 plays a role in biofilm formation, with luxS mutants showing a defect during biofilm formation and development27,34,35,36,37,38,39. The precise mode of action for LuxS in C. difficile has remained elusive as a result of conflicting studies and the lack of a clear receptor for AI-234,40,41.
Here we investigate the role of LuxS within C. difficile and mixed biofilm communities. Interestingly, we find that C. difficile LuxS/AI-2 mediates the induction of two putative C. difficile R20291 prophages within C. difficile biofilms. In mixed biofilms, we show that in the presence of B. fragilis, a gut bacterium, C. difficile growth is inhibited and this inhibition is alleviated in the absence of LuxS. Dual species transcriptomics show that distinct metabolic pathways are triggered in mixed cultures with the wild type (WT) and luxS mutant C. difficile strains.
Results
LuxS mediates biofilm formation in vitro
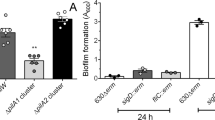
We previously reported that a R20291 C. difficile luxS mutant (luxS) was defective in biofilm formation as measured by crystal violet (CV) staining27. However, in our subsequent studies, although we see a reduction in biofilms at 24 h, we observe a high variability in the WT biofilms formed at 24 h and in the reduction in the luxS mutant between experiments (Fig. 1A). Nevertheless, the biofilm defect for luxS was very consistent at later time points (72 h) (Fig. 1A,B). In spite of differences in total biofilm content, colony counts from the WT and luxS biofilms were similar at both time points (Fig. 1C). There were also no differences in the spore content of WT and luxS biofilms (~0.003% at 24 h and ~0.1% at 72 h) (Fig. S1).

LuxS biofilm defect is reversed by addition of DPD. (A) WT and LuxS biofilms were grown for 24 h or 72 h and stained with 0.2% CV, followed by measuring OD570, N = 5. (B) Representative pictures of crystal violet stained C. difficile WT and luxS biofilms after 72 h. (C) Colony counts (vegetative cells) from biofilms (N = 7) after 24 h and 72 h. (D) The AI-2 precursor, DPD, was exogenously supplemented to LuxS at a concentration of 100 nM, followed by biofilm staining and quantitation with 0.2% CV after 72 h (N = 4). Error bars indicate SD, *p < 0.05, **p < 0.01, ***p < 0.001 as determined by Student’s t-test or by Mann-Whitney U test, ns- not significant.
To determine if AI-2 signalling is involved in biofilm formation, we first performed an AI-2 assay from both planktonic and biofilm supernatants as described by Carter et al. 2005 (Fig. S2A). AI-2 is produced maximally in mid-log and stationary phases as previously reported40. The WT strain produced less AI-2 in 24 h biofilms compared to log phase culture, while the luxS strain did not produce AI-2 as expected (Fig. S2B). To study if the reduction in biofilm formation in luxS could be complemented by chemically synthesised 4,5-dihydroxy-2, 3-pentanedione (DPD), the precursor of AI-2, was supplemented in the culture medium. Whilst high concentrations (>1000 nM) appeared to have only a partial effect on biofilm formation (Fig. S3), a concentration of 100 nM was capable of restoring the WT phenotype (Fig. 1D), indicating that AI-2 may be involved in signalling within C. difficile biofilms.
RNA-seq analysis reveals LuxS-mediated prophage induction
To investigate mechanism of action of LuxS/AI-2 in C. difficile, transcriptional profiles of C. difficile WT and luxS strains planktonically cultured (OD600 0.8 in BHI) were first compared using RNA-seq. However, surprisingly, no differential transcriptional changes were observed (Accession number E-MTAB-7486). Following this, an RNA-seq analysis was performed with total RNA isolated from C. difficile WT and luxS biofilms grown in BHIS +0.1 M glucose [(BHIS-G) conditions used for biofilm assays in Fig. 1] for 18 h (Accession number E-MTAB-7523). Both strains show similar planktonic culture growth rates in BHIS-G as reported previously27.
The DESeq2 variance analysis package42 was used to identify genes that were differentially expressed in luxS ≥1.6-fold relative to the WT strain, with a p -adjusted value ≤0.05. This pairwise analysis identified 21 differentially expressed genes (Fig. 2A,C) (Table 1). Interestingly, all 18 down-regulated genes correspond to two prophage regions located within the C. difficile R20291 genome, CDR20291_1415–1464 and CDR20291_1197–1226 (Fig. 2B), as identified using the online phage search tool, Phaster43,44. A Fisher’s exact test (p-value < 0.001 for both prophage regions) further confirmed an enrichment of differently regulated genes in prophage regions. There were only three genes upregulated in the luxS biofilms compared to the WT; two of these were involved in trehalose utilisation, while the third was a phosphotransferase system glucose-specific transporter.

Down-regulation of prophage genes in the C. difficile luxS mutant. (A) Pairwise analysis identified 21 differentially expressed genes in luxS (red points). All 18 down-regulated genes clustered into two regions. (B) Three prophage regions are identified in the C. difficile genome using Phaster. Regions 2 and 3 were down regulated in luxS. (C) Heat map representation of the genes that were differentially expressed in luxS, red and green indicate down- and up-regulation respectively when compared to WT. 18 prophage genes were found to be down-regulated in luxS relative to WT, whilst two genes involved in trehalose metabolism were up-regulated in luxS relative to WT. Data shown is the mean of 3 independent experiments in triplicates. Differential expression was defined as ≥1.6-fold change relative to WT with an adjusted p-value ≤ 0.05.
To demonstrate the presence of phage in the biofilm, cell-free supernatants were treated with DNase, before a subsequent DNA extraction was performed. As the bacterial cells were already removed, only DNA within intact bacteriophages would be protected from DNase. A 16S rRNA gene PCR was performed to confirm digestion of all free extracellular genomic DNA from the biofilm (Fig. 3A). PCRs with primers corresponding to genes specific to each prophage, confirmed that the DNA extracted had come from the phage (Fig. 3B). Since cell lysis is linked to phage release, we quantified and compared the total extracellular DNA (eDNA) content of luxS mutant and WT biofilms. The WT biofilms contained more eDNA compared to the luxS mutant at 24 h and 72 h (Fig. 3C). Overall, these data suggest that AI-2 may play a role in inducing prophages in C. difficile biofilms, which leads to phage-mediated host cell lysis and eDNA generation, which may contribute to subsequent biofilm accumulation.

Presence of phage and eDNA in C. difficile biofilms. The phage origin of DNA isolated from WT biofilms was confirmed by PCR, using primers for 16S (A) and two phage genes (CDR20291_1436 and CDR20291_1208) (B). The negative controls were run on a different part of the same gel. The gel pictures were trimmed with no adjustment to the intensities. WT-1–3 are three biological replicates. (C) Total eDNA extracted from the WT and luxS mutant biofilms after 24 h and 72 h, normalised to the biofilm biomass. N = 3, ****p < 0.0001 as determined by Mann-Whitney U test.
C. difficile is inhibited when cultured with B. fragilis in mixed biofilms
Given the high microbial density within the gut, C. difficile likely needs to interact with other members of the gut microbiota to establish itself within this niche. As an inter-species signalling function has been previously proposed for AI-245, we sought to investigate the interactions between a gut-associated Bacteroides spp and C. difficile. We examined C. difficile interactions with Bacteriodes fragilis, a gut commensal and pathogen, that has been previously reported to negatively correlate with CDI infections46.
C. difficile formed significantly more biofilms in vitro compared with B. fragilis in monocultures, as measured by CV staining (Fig. 4A). When both organisms were co-cultured, less biofilm was formed compared to C. difficile monoculture (Fig. 4A). Both B. fragilis and C. difficile grow well, although with slightly different growth rates, in BHIS-G (Fig. S4). To investigate the impact of co-culturing on both C. difficile and B. fragilis, bacterial numbers (CFU/ml) were determined from monoculture and mixed biofilms (Fig. 4B). Colony counts obtained from the mono and co-culture biofilms confirmed that B. fragilis was a poor biofilm producer when cultured alone. Interestingly, when both species were co-cultured, the CFU/ml for C. difficile was significantly reduced, and the CFU/ml of B. fragilis was significantly higher. This reduction of colony counts of C. difficile was observed at both 24 h (Fig. 4B) and 72 h (Fig. S5). AI-2 production from single and mixed biofilms was also quantitated. We observed no production of AI-2 by B. fragilis, and a reduction of AI-2 production by the mixed biofilms compared with the WT C. difficile biofilms (Fig. S6). C. difficile spore measurements from the mixed biofilms indicate that while the percentage of spores is higher (2%) due to the decrease in C. difficile numbers, there was no increase in the actual spore numbers (Fig. S7). These data suggest that the presence of B. fragilis in biofilms results in inhibition of C. difficile growth.

B. fragilis mediated inhibition of C. difficile in mixed biofilms. (A) Biofilm of C. difficile, B. fragilis and both species co-cultured (mixed) were grown for 24 h and stained with 0.2% CV, followed by measuring OD570. (B) Colony counts for both C. difficile (vegetative cells) and B. fragilis from mono and co-culture biofilms after 24 h. (C) Colony counts for both C. difficile (vegetative cells) and B. fragilis from mono and co-culture during planktonic growth. Data shown is the mean of 3 independent experiments in triplicates and error bars indicate SD, **p < 0.005, ****p < 0.0001 as determined by one-way ANOVA, Tukey’s multiple comparison test, ns -not significant (significant differences were determined for C. difficile or B. fragilis mean CFU counts between single and mixed biofilms).
To understand if the inhibitory effect was due to a factor secreted by B. fragilis, C. difficile and B. fragilis were co-cultured under planktonic conditions for 6 h and 10 h (Fig. 4C). However, there were no significant differences in C. difficile bacterial numbers between mono and co-culture. Additionally, supplementing biofilms with B. fragilis planktonic or biofilm culture supernatants did not cause C. difficile growth inhibition (Fig. S8), indicating that the observed inhibitory effects were specific to adherent biofilms i.e. when they are in close proximity to each other.
LuxS is involved in the B. fragilis inhibition of C. difficile
To study the role of LuxS in C. difficile-B. fragilis interactions, WT C. difficile and luxS strains were co-cultured with B. fragilis in mixed biofilms. CV staining of biofilms showed that there was less luxS biofilm formed compared to the WT when co-cultured with B. fragilis (Fig. 5A). While colony counts of C. difficile in monocultures were similar for both WT and luxS strains (Fig. 1B), when co-cultured with B. fragilis the bacterial counts for both C. difficile strains were significantly reduced, although the reduction was significantly higher for the WT than luxS (Fig. 5B). Colony counts for B. fragilis increased significantly during co-culture, with similar levels observed in both co-culture conditions (Fig. 5B). There was no increase in the spore numbers in the luxS in mixed biofilms (Fig. S7). These data suggest that AI2/LuxS is involved in mediating the B. fragilis-induced inhibition of C. difficile, when they are within adherent communities.

B. fragilis-mediated inhibition of C. difficile is more prominent for WT than LuxS. (A) Biofilms for mono and co-cultures of C. difficile WT and luxS with B. fragilis were grown for 24 h and stained with 0.2% CV and were quantified using a spectrophotometer OD570. (B) Colony counts for C. difficile WT, C. difficile luxS (vegetative cells) during co-culture with B. fragilis were performed at 24 h. Data shown is the mean of 3 independent experiments in triplicates and error bars indicate SD, **p < 0.01, ***p < 0.001 as determined by one-way ANOVA, Tukeys multiple comparison test (significant differences were determined for C. difficile or B. fragilis mean CFU counts between single and mixed biofilms).
Dual species RNA-seq analysis shows altered metabolism in C. difficile and B. fragilis in the absence of LuxS
To investigate mechanisms underlying the C. difficile inhibition mediated by B. fragilis, we performed an RNA-seq analysis to compare biofilm monocultures of C. difficile WT, luxS or B. fragilis with mixed biofilm co-cultures of C. difficile WT or luxS with B. fragilis (Accession number E-MTAB-7523). Differentially expressed genes were defined as having ≥1.6-fold relative to their respective control (mono-cultures of either B. fragilis or C. difficile WT), with an adjusted p-adjusted value ≤ 0.05.
We compared the expression profiles of C. difficile WT biofilms to C. difficile WT-B. fragilis mixed biofilms and C. difficile luxS biofilms to C. difficile luxS-B. fragilis mixed biofilms. A total of 45 genes were up-regulated (21) or down-regulated (24) in C. difficile WT (Fig. 6, Table 2), while 69 genes were differentially expressed in C. difficile luxS of which 34 were down-regulated and 35 up-regulated, during co-culture with B. fragilis (Fig. 6A, Table 3).

Dual species RNA-seq shows modulation of metabolic pathways in C. difficile WT, luxS and B. fragilis. Heat maps showing clustering of up- and down-regulated genes in (A) C. difficile WT and luxS co-cultured with B. fragilis compared to C. difficile WT mono-culture, and in (B) B. fragilis co-cultured with C. difficile WT and luxS compared to B. fragilis mono-culture. Red indicates genes that are down-regulated, whilst green indicates genes that are up-regulated.
Eight up-regulated genes were specific to C. difficile WT co-culture (Table 2). Of these, four genes involved in fatty acid biosynthesis and metabolism: fabH encoding 3-oxoacyl-[acyl-carrier protein] synthase III, fabK encoding trans-2-enoyl-ACP reductase, accC encoding a biotin carboxylase (acetyl-CoA carboxylase subunit A), and accB encoding a biotin carboxyl carrier protein of acetyl-CoA carboxylase (Table 2). 11 genes were down-regulated exclusively in WT however, these genes do not coincide with a specific metabolic pathway.
18 up-regulated genes were specific to luxS in co-culture (Table 3). These include a putative homocysteine S-methyltransferase, a putative osmoprotectant ABC transporter, substrate binding/ permease protein, ribonucleoside-diphosphate reductase alpha chain (nrdE) and two genes from the trehalose operon: a PTS system II ABC transporter, and trehalose-6-phosphate hydrolase (treA). 24 genes were down-regulated (Table 3), which include 3 genes involved in thiamine metabolism thiD, thiK and thiE1, (CDR20291_1497, CDR20291_1498 and CDR20291_1499 respectively) which encode a putative phosphomethylpyrimidine kinase, 4-methyl-5-beta-hydroxyethylthiazole kinase and thiamine-phosphate pyrophosphorylase respectively.
A total of 26 genes were differentially expressed in both C. difficile WT and C. difficile luxS when co-cultured with B. fragilis (Fig. 6A, Table S1). These include six up-regulated genes (accB, abfH, abfT, abfD, sucD and cat1) involved in carbon and butanoate metabolism, with cat1, which encodes succinyl-CoA:coenzyme A transferase, being the highest up-regulated gene for both C. difficile strains.
When C. difficile WT-B. fragilis mixed biofilms and luxS-B. fragilis mixed biofilms were compared to B. fragilis single biofilms, in contrast, a higher number of genes (266) were differentially expressed in B. fragilis when co-cultured with C. difficile (WT and luxS) (Table S4, Fig. 6B). A total of 114 B. fragilis genes were found to be specific to C. difficile WT co-culture, with 56 of these up-regulated and 58 down-regulated (Table S2). Similarly, 91 genes were found to be specific to C. difficile luxS co-culture, with 56 of these up-regulated and 35 down-regulated (Table S3). Although distinct B. fragilis expression profiles were observed with the WT and luxS (Fig. 6B), there were no clear pathways identified in the datasets.
Whilst the highest up-regulated gene in both WT and LuxS co-cultures encodes a putative virus attachment protein, no other viral genes were shown to be up-regulated (Table S4). Genes encoding iron containing proteins desulfoferrodoxin and rubrerythrin were highly up-regulated in both conditions. Interestingly, multiple copies of fecR, a key regulator for the ferric citrate transport system47, and ferrous iron transport protein B were also up-regulated. Additionally, a number of other metabolic pathways were up-regulated, including four genes (encoding 3-isopropylmalate dehydratase small subunit, 3-isopropylmalate dehydrogenase, Galactokinase and 3-isopropylmalate dehydratase large subunit) involved in valine, leucine and isoleucine biosynthesis and C5-branched dibasic acid metabolism. It should be noted that many of the up-regulated genes were hypothetical proteins of unknown function. Similarly, several metabolic pathways were down-regulated in both co-culture conditions. These include six genes involved in carbon metabolism, four genes involved in alanine, aspartate and glutamate metabolism, and four genes involved in the biosynthesis of amino acids, although these appear to be single genes rather than specific pathways.
Discussion
Inter-bacterial interactions within gut communities are critical in controlling invasion by intestinal pathogens. Quorum sensing molecules such as AI-2 are instrumental in bacterial communication, especially during formation of bacterial communities31,36,37,48,49,50. C. difficile produces AI-2, although the mechanism of action of LuxS/AI-2 in C. difficile, particularly within a biofilm community is unclear. We report here that the C. difficile LuxS/AI-2 plays an important role in the formation of single and multi-species communities. In C. difficile biofilms, LuxS mediates the induction of prophages, which likely contributes to the biofilm structure. Whereas, in a mixed biofilm of C. difficile and the intestinal commensal and pathogen, B. fragilis, LuxS likely triggers the induction of differential metabolic responses in B. fragilis, that leads to growth inhibition of C. difficile. To our knowledge, this is the first time dual species RNA-seq51 has been applied to analyse interactions between anaerobic gut bacteria in an adherent biofilm community.
Bacterial biofilms contain a number of extracellular components that make up their complex structure including extracellular DNA (eDNA), a key component that binds together bacteria within a community. Autolysis is a common mechanism by which eDNA is released from bacterial cells52. In bacteria such as Staphylococcus aureus and Pseudomonas aeruginosa, eDNA is generated through the lysis of subpopulations within a biofilm, under the control of quorum sensing52,53,54,55. In C. difficile luxS mutant (luxS) biofilms, we observe reduced induction of two C. difficile prophages compared with the WT. These phage loci were conserved in several C. difficile strains with the Region 2 encoding a phiC2-like, phi-027 phage56,57,58. Given that it has previously been shown that eDNA is a major component of C. difficile biofilms27,30, it is likely that phage-mediated bacterial cell lysis and subsequent DNA release help build a biofilm. Indeed, given the detection of eDNA in the luxS biofilms, there are likely other unknown eDNA release mechanisms during C. difficile biofilm formation.
Phages are also known to control biofilm structure in some organisms: a filamentous phage of P. aeruginosa was reported to be a structural component of the biofilm59 and an AI-2 induced phage mediated the dispersal of Enterococcus faecalis biofilms60. Although attempts to visualise phages from C. difficile biofilms with transmission electron microscopy (data not shown) were unsuccessful, we cannot rule out the possibility that C. difficile phages may directly influence the biofilm structure. While the precise mechanisms by which LuxS/AI-2 controls phage induction are yet to be elucidated, AI-2 appears to be signalling through a yet unidentified AI-2 receptor in C. difficile. Phage-mediated control of biofilms may in part explain the variation observed in biofilm formation between different C. difficile strains61.
The human gut hosts a variety of bacterial species, which compete or coexist with each other. It is likely that the bacteria occupying this niche form multi-species bacterial communities in association with the mucus layer. Interactions within such communities are important in gaining a better understanding of phenomena such as ‘colonisation resistance’ which prevents pathogens such as C. difficile from establishing an infection14. Whilst sequencing studies have identified members of the Bacteroides genus as being associated with gut colonisation resistance to C. difficile, the mechanisms have remained elusive10. A recent study demonstrated that production of the enzyme: bile salt hydrolase, is responsible for the inhibitory effect of B. ovatus on C. difficile62. This study reported that in the presence of bile acids, cell free supernatants for B. ovatus were capable of inhibiting the growth of C. difficile whereas in the absence of bile acids, C. difficile growth was promoted. Since bile acids are not supplemented into our media, a different mechanism is likely responsible for B. fragilis mediated inhibition of C. difficile. Also, the growth restraining effects of B. fragilis on C. difficile were evident only within mixed biofilms, not in planktonic culture or with culture supernatants. While it is likely that cell-cell contact is essential for the inhibitory effect, we cannot exclude involvement of an inhibitory secreted molecule that accumulates to a higher concentration within a biofilm environment, or that B. fragilis has a competitive growth advantage in a biofilm environment.
A dual species RNA-seq analysis performed to understand the interactions between the two bacterial species, showed that largely all the differentially expressed genes mapped to distinct metabolic pathways. Overall, a higher number of genes were modulated in B. fragilis as compared to C. difficile strain during co-culture, which is in line with the growth characteristics observed. Carbon and butanoate metabolism pathways were induced in C. difficile strains in response to co-culture (accB, abfH, abfT, abfD, sucD and cat1). An up-regulation of the succinate utilisation genes was also recently reported in C. difficile 630 microfermenter biofilm cells as compared to planktonic cells63. As B. fragilis is known to produce succinate64, it is likely that the upregulation in these pathways results from the increased levels of succinate in the culture medium. However, since gut microbiota-produced succinate promotes C. difficile growth in vivo17, it is unlikely that these changes are directly responsible for the observed inhibition of C. difficile. However, bacteria utilise carbohydrates in a sequential manner65. Consistent with this, we observed a down-regulation of genes important for the utilisation of pyruvate such as bcd2 and idhA encoding for butyryl-CoA dehydrogenase and (r)-2-hydroxyisocaproate dehydrogenase respectively. A down-regulation of sugar fermentation pathway genes was also observed by Poquet et al. in single species C. difficile biofilms compared to planktonic cultures63. Such a shift in metabolism could allow B. fragilis to fully consume other metabolites, and thus enabling it to outcompete C. difficile.
Additionally, it is interesting to note that a number of copies of the ferric citrate transport system regulator, fecR, are up-regulated in B. fragilis during C. difficile co-culture. The ferric citrate transport system is an iron uptake system that responds to the presence of citrate47,66. Analysis of the C. difficile genome using BLAST (NCBI) showed that C. difficile does not possess this iron uptake system. Given the evidence that ferric citrate is an iron source in the gut47,67, B. fragilis may have an advantage over C. difficile in sequestering iron, and thus preventing C. difficile colonisation. Although the clear modulation of metabolic pathways strongly suggest a competitive advantage of B. fragilis over C. difficile, it is possible that the genes with unknown functions that are differentially expressed in B. fragilis (Table S1), encode pathways for the production of yet to be identified small inhibitory molecules.
The LuxS/AI-2 quorum sensing system is known to have a cross-species signalling role in many bacteria45,68. While all sequenced C. difficile strains produce AI-2, only selected strains of B. fragilis have the ability to produce AI-269. A recent study showed that Ruminococcus obeuem inhibited Vibrio cholerae in the gut via LuxS/AI-2 mediated downregulation of V. cholerae colonisation factors70. Also, AI-2 produced by engineered E. coli was reported to influence firmicutes/bacteroidetes ratios in microbiota treated with streptomycin71. Our data show the involvement of LuxS/AI-2 in the B. fragilis-mediated C. difficile growth inhibition. As the B. fragilis strain used in this study does not produce AI-2, it is likely that B. fragilis responds differentially to AI-2 produced by C. difficile. Similar to the WT, most transcriptional changes were in metabolic pathways, although specific sets of genes were modulated in the luxS mutant. Modulation of prophage genes was not seen, unlike single biofilm cultures, indicating different dominant mechanisms at play in a multi-species environment.
It was interesting to note that the trehalose utilisation operon, which provides a growth advantage to C. difficile against other gut bacteria72, was upregulated in both the single luxS biofilms (Table 1) and luxS co-cultured with B. fragilis (Table 3). The upregulation of a phosphotransferase system component and treA (both in the same operon), likely enables increased utilisation of trehalose, providing an additional carbon source. Like glucose, trehalose acts as an osmoprotectant73 and its presence within the cell may help maintain protein conformation during cellular dehydration. It is possible that trehalose plays a role in building C. difficile biofilms, as reported for Candida biofilms74. Our preliminary studies with exogenous trehalose levels similar to those used by Collins et al. (2018) showed an inhibitory effect on biofilm formation by both WT C. difficile and luxS, although no differential effects were observed between luxS and WT (Fig. S9). However, further investigations into accumulation of trehalose within biofilms are required to clarify the role of trehalose in luxS mediated biofilm formation.
In conclusion, we report that C. difficile LuxS/AI-2 may play a key role in building C. difficile communities through mediating prophage induction, and subsequent accumulation of eDNA. In mixed communities, C. difficile AI-2 likely signals to B. fragilis to induce an altered metabolic response, enabling it to outgrow C. difficile. Further studies are required to understand the precise AI-2 sensing pathways involved.
Materials and Methods
Bacterial strains and media
Two bacterial species were used in this study – C. difficile strain: B1/NAP1/027 R20291 (isolated from the Stoke Mandeville outbreak in 2004 and 2005), and Bacteroides fragilis (a clinical isolate from a biliary stent kindly provided by Dr Claudia Vuotto and Dr Gianfranco Donelli, Rome). A luxS Clostron R20291 mutant described previously in Dapa et al.27 was used in this study. Both species were cultured under anaerobic conditions (80% N2, 10% CO2, 10% H2) at 37 °C in an anaerobic workstation (Don Whitley, United Kingdom) in BHIS, supplemented with L-Cysteine (0.1% w/v; Sigma, United Kingdom), yeast extract (5 g/l; Oxoid) and glucose (0.1 M).
Vibrio harveyi strain: BB170 was used to measure AI-2. V. harveyi strains were cultured in aerobic conditions at 30 °C in Lysogeny broth (LB) supplemented with kanamycin (50 µg/ml).
Biofilm formation assay
Biofilms were grown as per the previously published protocol27. Overnight cultures of C. difficile were diluted 1:100 in fresh BHIS with 0.1 M glucose. 1 ml aliquots were pipetted into 24-well tissue culture treated polystyrene plates (Costar), and incubated under anaerobic condition at 37 °C, for 6–120 h. Tissue culture plates were pre-incubated for 48 h prior to use. The plates were wrapped with parafilm to prevent liquid evaporation.
Measurement of biofilm biomass
Biofilm biomass was measured using crystal violet (CV)27. After the required incubation, each well of the 24-well plate was washed with sterile phosphate buffer saline (PBS) and allowed to dry for a minimum of 10 mins. The biofilm was stained using 1 ml of filter-sterilised 0.2% CV and incubated for 30 mins at 37 °C, in anaerobic conditions. The CV was removed from each well, and wells were subsequently washed twice with sterile PBS. The dye was extracted by incubated with 1 ml methanol for 30 mins at room temperature in aerobic conditions. The methanol-extracted dye was diluted 1:1, 1:10 or 1:100 and OD570 was measured with a spectrophotometer (BMG Labtech, UK).
For bacterial cell counts from the biofilm, the planktonic phase was removed and wells were washed once using sterile PBS. The adherent biofilms were then detached by scrapping with a sterile pipette tip and re-suspended into 1 ml PBS. Serial dilutions were made and plated onto BHIS plates to determine the CFU present in the biofilm.
Co-culture biofilm assay
For generation of co-culture biofilms, both C. difficile and B. fragilis were diluted to an OD600 of 1. Both species were diluted 1:100 into fresh BHIS with 0.1 M glucose. Biofilms assays were performed as described above and measured by a combination of CV staining and CFU. To distinguish between C. difficile and B. fragilis, serial dilutions used for determining CFU were plated on BHIS plates additionally supplemented with C. difficile selective supplement (Oxoid, UK). Colonies can be differentiated by size and colony morphology as B. fragilis form very small colonies.
Exogenous addition of DPD
To analyse the potential signalling role of AI-2, biofilm assays were performed as described above in BHIS with 0.1 M glucose containing 1 nM, 10 nM, 100 nM, or 1 µM of chemically synthesised, exogenous 4,5-Dihydroxy-2,3-pentanedione (Omm Scientific, Texas USA) for both C. difficile WT and LuxS. BHIS with 0.1 M glucose was used as a control. Samples were washed and stained with 0.2% CV at either 24 h or 72 h.
AI-2 Assay
The AI-2 bioluminescence assay was carried out essentially as described by Bassler et al. 199375. The V. harveyi reporter strain BB170 was grown overnight in LB medium before being diluted 1: 5000 in Autoinducer Bioassay (AB) medium containing 10% (v/v) cell-free conditioned medium collected from either planktonic or biofilm C. difficile cultures (in BHI) and allowed to grow at 30 °C with shaking. AB medium containing 10% (v/v) from V. harveyi BB120 was used as a positive control, and 10% (v/v) sterile BHI medium as a blank. Luminescence was measured every hour using a SPECTROstar Omega plate reader. Induction of luminescence was taken at the time when there was maximal difference between the positive and negative controls (usually 2–5 h) and is expressed as a percentage of the induction observed in the positive control.
RNA-seq
Biofilms were grown for 18 h in BHIS + glucose, supernatants were removed and attached biofilms were washed with 1 ml PBS. Biofilms were disrupted and RNA was extracted using Trizol (Invitrogen, UK). 5 µg of extracted RNA was treated with RiboZEROTM (Illumina, UK) according to the manufacturer’s protocol to deplete rRNA. cDNA libraries were prepared using TruSEQ LT (Illumina, UK) according to the manufacturer’s instructions. Briefly, samples were end-repaired, mono-adenylated, ligated to index/adaptors. Libraries were quantified by bioanalyzer and fluorometer assay. The final cDNA library was prepared to a concentration of 10–12 pM and sequenced using paired end technology using a version-3 150-cycle kit on an Illumina MiSeqTM (Illumina, UK).
RNA-seq analysis
The paired-end sequencing reads from RNA-seq experiments were mapped against the appropriate reference genome (NC_013316 for C. difficile and a de novo assembly using RNA SPAdes v3.9 with default settings76 from the RNA-sequence reads for B. fragilis [Accession number PRJEB29695). The first read was flipped into the correct orientation using seqtk v1.3 (https://github.com/lh3/seqtk) and the reads were mapped against the reference genome using BWA v0.7.5 with the ‘mem’ alignment algorithm77. BAM files were manipulated with Samtools v0.1.18 using the ‘view’ and ‘sort’ settings77. Sorted BAM and GFF (general feature format) files were inputted into the coverageBed tool v2.27.0 with default settings78 to gain abundance of each genomic feature. The R package DESeq2 was used with default settings to calculate differential gene expression using a negative binomial distribution model42. The data was filtered by applying a cut-off of 1.6 for the fold change and 0.05 for the adjusted p-value. All sequencing reads were submitted to the European Bioinformatics Institute (Accession numbers E-MTAB-7486, E-MTAB-7523 and PRJEB29695).
As analysis by BLAST (NCBI) demonstrated species specificity for mapping, co-culture samples were mapped to each species reference separately. Initial mapping of the B. fragilis strain to a published reference proved unsuccessful, offering a poor rate of alignment of 60%. As the B. fragilis strain has not been previously sequenced, and because we were not successful in generating high quality genome sequence, a reference was generated from RNA library of B. fragilis using the software rnaSPAdes v3.979 and annotated using Prokka v1.11 (default settings)80. The reads from each condition were mapped to their respective reference sequence using BWA v0.7.5 (‘mem’ algorithm)77,81 and counted using coverageBed v2.27.078. Metabolic pathways in C. difficile were identified using the KEGG mapper82, a tool that identifies the function of genes in a published genome. As the B. fragilis strain used in this study does not have a published reference genome, blastKOALA83 was used to search for gene homology within metabolic pathways. Heatmaps were generated from normalised gene expression data outputted from DESeq2, using the online tool Heatmapper84 using the default settings.
PCR analysis
16S PCRs were performed using the universal 16S rRNA bacterial primers 27F and 1392R (Table S2). Primers were constructed for prophage genes CDR20219_1208 and CDR20291_1436 (Table S2) to confirm the presence of prophage within C. difficile biofilms. PCR was carried out using Fusion High-Fidelity DNA polymerase (NEB, USA) following the manufacturer’s protocol. Samples were heated to 95 °C for 5 mins followed by 35 cycles of: 95 °C for 30 seconds, 51 °C for 30 seconds and 72 °C for 30 seconds, after which samples were heated to 72 °C for 10 mins.
eDNA quantification
eDNA was extracted from C. difficile biofilms grown in a 24-well plate as described above, using a protocol described in Rice et al.53. Briefly, the plate was sealed with parafilm and chilled at 4 °C for 1 hour. 1 μl 0.5 M EDTA was added to each well and incubated at 4 °C for 5 mins. The medium was removed and biofilms were resuspended in 300 μl 50 mM TES buffer (50 mM Tris HCl/10 mM EDTA/500 mM NaCl). The OD600 was measured to determine biofilm biomass and the tubes were centrifuged at 4 °C at 18,000 g for 5 mins, and was used to normalise the eDNA values. 100 μl of supernatant was transferred to a tube of chilled TE buffer (10 mM Tris HCl/ 1 mM EDTA) on ice. DNA was extracted using an equal volume of phenol/chloroform/isoamyl alcohol three times. 3 volumes of ice-cold 100% ethanol and 1/10 volumes 3 M sodium acetate were added to the aqueous phase to precipitate the DNA. The DNA pellet was washed with 1 ml ice-cold 70% ethanol, dissolved in 20 μl TE buffer, quantified by Qubit fluorometer (Thermo Fisher).
Spore counts
To determine the number of spores, adherent biofilms were resuspended in PBS and treated at 65 °C for 25 mins as previously described85. Untreated and heat-treated samples were serial diluted and plated on BHIS and BHIS-TC agar (supplemented with 0.1% sodium taurocholate, Sigma-Aldrich, UK). No bacteria were obtained from the heat-treated samples plated on BHI (without sodium taurocholate). The CFU/ml obtained from heat-treated samples plated on BHIS-TC plates represent heat-resistant spores, and the CFU/ml obtained from untreated samples plated on BHIS plates represent the total cell counts.
Statistical analysis
All experiments were performed in triplicate, with at least three independent experiments performed. Paired or unpaired student’s t-test was used to determine if differences between two groups were significant, and one way-ANOVA was used to compare multiple groups. Mann-Whitney U tests were used to compare non-parametric data. Fisher’s exact t-test was used to confirm the enrichment of differently regulated genes in prophage regions.
References
Lessa, F. C. et al. Burden of Clostridium difficile infection in the United States. N Engl J Med 372, 825–834, https://doi.org/10.1056/NEJMoa1408913 (2015).
Davies, K. A. et al. Underdiagnosis of Clostridium difficile across Europe: the European, multicentre, prospective, biannual, point-prevalence study of Clostridium difficile infection in hospitalised patients with diarrhoea (EUCLID). Lancet Infect Dis 14, 1208–1219, https://doi.org/10.1016/S1473-3099(14)70991-0 (2014).
Gravel, D. et al. Health care-associated Clostridium difficile infection in adults admitted to acute care hospitals in Canada: a Canadian Nosocomial Infection Surveillance Program Study. Clin Infect Dis 48, 568–576, https://doi.org/10.1086/596703 (2009).
Collins, D. A., Selvey, L. A., Celenza, A. & Riley, T. V. Community-associated Clostridium difficile infection in emergency department patients in Western Australia. Anaerobe 48, 121–125, https://doi.org/10.1016/j.anaerobe.2017.08.008 (2017).
Barbut, F. et al. Epidemiology of recurrences or reinfections of Clostridium difficile-associated diarrhea. J Clin Microbiol 38, 2386–2388 (2000).
Rupnik, M., Wilcox, M. H. & Gerding, D. N. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7, 526–536, https://doi.org/10.1038/nrmicro2164 (2009).
Smits, W. K., Lyras, D., Lacy, D. B., Wilcox, M. H. & Kuijper, E. J. Clostridium difficile infection. Nat Rev Dis Primers 2, 16020, https://doi.org/10.1038/nrdp.2016.20 (2016).
Hopkins, M. J. & Macfarlane, G. T. Changes in predominant bacterial populations in human faeces with age and with Clostridium difficile infection. J Med Microbiol 51, 448–454, https://doi.org/10.1099/0022-1317-51-5-448 (2002).
Manges, A. R. et al. Comparative metagenomic study of alterations to the intestinal microbiota and risk of nosocomial Clostridum difficile-associated disease. J Infect Dis 202, 1877–1884, https://doi.org/10.1086/657319 (2010).
Seekatz, A. M. et al. Recovery of the gut microbiome following fecal microbiota transplantation. mBio 5, e00893–00814, https://doi.org/10.1128/mBio.00893-14 (2014).
Schubert, A. M. et al. Microbiome data distinguish patients with Clostridium difficile infection and non-C. difficile-associated diarrhea from healthy controls. mBio 5, e01021–01014, https://doi.org/10.1128/mBio.01021-14 (2014).
Khoruts, A., Dicksved, J., Jansson, J. K. & Sadowsky, M. J. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J Clin Gastroenterol 44, 354–360, https://doi.org/10.1097/MCG.0b013e3181c87e02 (2010).
Hamilton, M. J., Weingarden, A. R., Unno, T., Khoruts, A. & Sadowsky, M. J. High-throughput DNA sequence analysis reveals stable engraftment of gut microbiota following transplantation of previously frozen fecal bacteria. Gut Microbes 4, 125–135, https://doi.org/10.4161/gmic.23571 (2013).
Britton, R. A. & Young, V. B. Role of the intestinal microbiota in resistance to colonization by Clostridium difficile. Gastroenterology 146, 1547–1553, https://doi.org/10.1053/j.gastro.2014.01.059 (2014).
Sorg, J. A. & Sonenshein, A. L. Bile salts and glycine as cogerminants for Clostridium difficile spores. J Bacteriol 190, 2505–2512, https://doi.org/10.1128/JB.01765-07 (2008).
Buffie, C. G. et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517, 205–208, https://doi.org/10.1038/nature13828 (2015).
Ferreyra, J. A. et al. Gut microbiota-produced succinate promotes C. difficile infection after antibiotic treatment or motility disturbance. Cell Host Microbe 16, 770–777, https://doi.org/10.1016/j.chom.2014.11.003 (2014).
Ng, K. M. et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature 502, 96–99, https://doi.org/10.1038/nature12503 (2013).
Kuehne, S. A. et al. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467, 711–713, https://doi.org/10.1038/nature09397 (2010).
Lyras, D. et al. Toxin B is essential for virulence of Clostridium difficile. Nature 458, 1176–1179, https://doi.org/10.1038/nature07822 (2009).
Voth, D. E. & Ballard, J. D. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18, 247–263, https://doi.org/10.1128/CMR.18.2.247-263.2005 (2005).
Merrigan, M. M. et al. Surface-layer protein A (SlpA) is a major contributor to host-cell adherence of Clostridium difficile. PloS one 8, e78404, https://doi.org/10.1371/journal.pone.0078404 (2013).
Waligora, A. J. et al. Characterization of a cell surface protein of Clostridium difficile with adhesive properties. Infect Immun 69, 2144–2153, https://doi.org/10.1128/IAI.69.4.2144-2153.2001 (2001).
Kovacs-Simon, A. et al. Lipoprotein CD0873 is a novel adhesin of Clostridium difficile. J Infect Dis 210, 274–284, https://doi.org/10.1093/infdis/jiu070 (2014).
Barketi-Klai, A., Hoys, S., Lambert-Bordes, S., Collignon, A. & Kansau, I. Role of fibronectin-binding protein A in Clostridium difficile intestinal colonization. J Med Microbiol 60, 1155–1161, https://doi.org/10.1099/jmm.0.029553-0 (2011).
Tasteyre, A., Barc, M.-C., Collignon, A., Boureau, H. & Karjalainen, T. Role of FliC and FliD Flagellar Proteins of Clostridium difficile in Adherence and Gut Colonization. Infection and Immunity 69, 7937–7940, https://doi.org/10.1128/iai.69.12.7937-7940.2001 (2001).
Dapa, T. et al. Multiple factors modulate biofilm formation by the anaerobic pathogen Clostridium difficile. J Bacteriol 195, 545–555, https://doi.org/10.1128/JB.01980-12 (2013).
Dawson, L. F., Valiente, E., Faulds-Pain, A., Donahue, E. H. & Wren, B. W. Characterisation of Clostridium difficile biofilm formation, a role for Spo0A. PloS one 7, e50527, https://doi.org/10.1371/journal.pone.0050527 (2012).
Mathur, H., Rea, M. C., Cotter, P. D., Hill, C. & Ross, R. P. The efficacy of thuricin CD, tigecycline, vancomycin, teicoplanin, rifampicin and nitazoxanide, independently and in paired combinations against Clostridium difficile biofilms and planktonic cells. Gut Pathog 8, 20, https://doi.org/10.1186/s13099-016-0102-8 (2016).
Semenyuk, E. G. et al. Spore formation and toxin production in Clostridium difficile biofilms. PloS one 9, e87757, https://doi.org/10.1371/journal.pone.0087757 (2014).
Li, Y. H. & Tian, X. Quorum sensing and bacterial social interactions in biofilms. Sensors (Basel) 12, 2519–2538, https://doi.org/10.3390/s120302519 (2012).
Antunes, L. C., Ferreira, R. B., Buckner, M. M. & Finlay, B. B. Quorum sensing in bacterial virulence. Microbiology 156, 2271–2282, https://doi.org/10.1099/mic.0.038794-0 (2010).
Vendeville, A., Winzer, K., Heurlier, K., Tang, C. M. & Hardie, K. R. Making ‘sense’ of metabolism: autoinducer-2, LuxS and pathogenic bacteria. Nat Rev Microbiol 3, 383–396, https://doi.org/10.1038/nrmicro1146 (2005).
Rezzonico, F. & Duffy, B. Lack of genomic evidence of AI-2 receptors suggests a non-quorum sensing role for luxS in most bacteria. BMC Microbiol 8, 154, https://doi.org/10.1186/1471-2180-8-154 (2008).
Auger, S., Krin, E., Aymerich, S. & Gohar, M. Autoinducer 2 affects biofilm formation by Bacillus cereus. Appl Environ Microbiol 72, 937–941, https://doi.org/10.1128/AEM.72.1.937-941.2006 (2006).
De Araujo, C., Balestrino, D., Roth, L., Charbonnel, N. & Forestier, C. Quorum sensing affects biofilm formation through lipopolysaccharide synthesis in Klebsiella pneumoniae. Res Microbiol 161, 595–603, https://doi.org/10.1016/j.resmic.2010.05.014 (2010).
Hardie, K. R. & Heurlier, K. Establishing bacterial communities by ‘word of mouth’: LuxS and autoinducer 2 in biofilm development. Nat Rev Microbiol 6, 635–643, https://doi.org/10.1038/nrmicro1916 (2008).
Huang, Z. et al. luxS-based quorum-sensing signaling affects Biofilm formation in Streptococcus mutans. J Mol Microbiol Biotechnol 17, 12–19, https://doi.org/10.1159/000159193 (2009).
Karim, M. M. et al. LuxS affects biofilm maturation and detachment of the periodontopathogenic bacterium Eikenella corrodens. J Biosci Bioeng 116, 313–318, https://doi.org/10.1016/j.jbiosc.2013.03.013 (2013).
Carter, G. P., Purdy, D., Williams, P. & Minton, N. P. Quorum sensing in Clostridium difficile: analysis of a luxS-type signalling system. J Med Microbiol 54, 119–127 (2005).
Lee, A. S. & Song, K. P. LuxS/autoinducer-2 quorum sensing molecule regulates transcriptional virulence gene expression in Clostridium difficile. Biochem Biophys Res Commun 335, 659–666, https://doi.org/10.1016/j.bbrc.2005.07.131 (2005).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550, https://doi.org/10.1186/s13059-014-0550-8 (2014).
Arndt, D. et al. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44, W16–21, https://doi.org/10.1093/nar/gkw387 (2016).
Zhou, Y., Liang, Y., Lynch, K. H., Dennis, J. J. & Wishart, D. S. PHAST: a fast phage search tool. Nucleic Acids Res 39, W347–352, https://doi.org/10.1093/nar/gkr485 (2011).
Bassler, B. L., Greenberg, E. P. & Stevens, A. M. Cross-species induction of luminescence in the quorum-sensing bacterium Vibrio harveyi. J Bacteriol 179, 4043–4045 (1997).
Goldberg, E. et al. The correlation between Clostridium-difficile infection and human gut concentrations of Bacteroidetes phylum and clostridial species. Eur J Clin Microbiol Infect Dis 33, 377–383, https://doi.org/10.1007/s10096-013-1966-x (2014).
Andrews, S. C., Robinson, A. K. & Rodriguez-Quinones, F. Bacterial iron homeostasis. FEMS Microbiol Rev 27, 215–237, https://doi.org/10.1016/S0168-6445(03)00055-X (2003).
Zhu, J. & Mekalanos, J. J. Quorum sensing-dependent biofilms enhance colonization in Vibrio cholerae. Dev Cell 5, 647–656 (2003).
Sakuragi, Y. & Kolter, R. Quorum-sensing regulation of the biofilm matrix genes (pel) of Pseudomonas aeruginosa. J Bacteriol 189, 5383–5386, https://doi.org/10.1128/JB.00137-07 (2007).
Solano, C., Echeverz, M. & Lasa, I. Biofilm dispersion and quorum sensing. Curr Opin Microbiol 18, 96–104, https://doi.org/10.1016/j.mib.2014.02.008 (2014).
Wolf, T., Kammer, P., Brunke, S. & Linde, J. Two’s company: studying interspecies relationships with dual RNA-seq. Curr Opin Microbiol 42, 7–12, https://doi.org/10.1016/j.mib.2017.09.001 (2018).
Ibanez de Aldecoa, A. L., Zafra, O. & Gonzalez-Pastor, J. E. Mechanisms and Regulation of Extracellular DNA Release and Its Biological Roles in Microbial Communities. Front Microbiol 8, 1390, https://doi.org/10.3389/fmicb.2017.01390 (2017).
Rice, K. C. et al. The cidA murein hydrolase regulator contributes to DNA release and biofilm development in Staphylococcus aureus. Proc Natl Acad Sci USA 104, 8113–8118, https://doi.org/10.1073/pnas.0610226104 (2007).
Brackman, G. et al. The Quorum Sensing Inhibitor Hamamelitannin Increases Antibiotic Susceptibility of Staphylococcus aureus Biofilms by Affecting Peptidoglycan Biosynthesis and eDNA Release. Sci Rep 6, 20321, https://doi.org/10.1038/srep20321 (2016).
Svensson, S. L., Pryjma, M. & Gaynor, E. C. Flagella-mediated adhesion and extracellular DNA release contribute to biofilm formation and stress tolerance of Campylobacter jejuni. PloS one 9, e106063, https://doi.org/10.1371/journal.pone.0106063 (2014).
Nale, J. Y. et al. Diverse temperate bacteriophage carriage in Clostridium difficile 027 strains. PloS one 7, e37263, https://doi.org/10.1371/journal.pone.0037263 (2012).
Stabler, R. A. et al. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol 10, R102, https://doi.org/10.1186/gb-2009-10-9-r102 (2009).
Sekulovic, O. & Fortier, L. C. Global transcriptional response of Clostridium difficile carrying the CD38 prophage. Appl Environ Microbiol 81, 1364–1374, https://doi.org/10.1128/AEM.03656-14 (2015).
Secor, P. R. et al. Biofilm assembly becomes crystal clear - filamentous bacteriophage organize the Pseudomonas aeruginosa biofilm matrix into a liquid crystal. Microb Cell 3, 49–52, https://doi.org/10.15698/mic2016.01.475 (2015).
Rossmann, F. S. et al. Phage-mediated dispersal of biofilm and distribution of bacterial virulence genes is induced by quorum sensing. PLoS Pathog 11, e1004653, https://doi.org/10.1371/journal.ppat.1004653 (2015).
Pantaleon, V. et al. Clostridium difficile forms variable biofilms on abiotic surface. Anaerobe, https://doi.org/10.1016/j.anaerobe.2018.05.006 (2018).
Yoon, S. et al. Bile salt hydrolase-mediated inhibitory effect of Bacteroides ovatus on growth of Clostridium difficile. J Microbiol 55, 892–899, https://doi.org/10.1007/s12275-017-7340-4 (2017).
Poquet, I. et al. Clostridium difficile Biofilm: Remodeling Metabolism and Cell Surface to Build a Sparse and Heterogeneously Aggregated Architecture. Front Microbiol 9, 2084, https://doi.org/10.3389/fmicb.2018.02084 (2018).
Macy, J. M., Ljungdahl, L. G. & Gottschalk, G. Pathway of succinate and propionate formation in Bacteroides fragilis. J Bacteriol 134, 84–91 (1978).
Bruckner, R. & Titgemeyer, F. Carbon catabolite repression in bacteria: choice of the carbon source and autoregulatory limitation of sugar utilization. FEMS Microbiol Lett 209, 141–148, https://doi.org/10.1111/j.1574-6968.2002.tb11123.x (2002).
Wagegg, W. & Braun, V. Ferric citrate transport in Escherichia coli requires outer membrane receptor protein fecA. J Bacteriol 145, 156–163 (1981).
Kortman, G. A., Raffatellu, M., Swinkels, D. W. & Tjalsma, H. Nutritional iron turned inside out: intestinal stress from a gut microbial perspective. FEMS Microbiol Rev 38, 1202–1234, https://doi.org/10.1111/1574-6976.12086 (2014).
Federle, M. J. & Bassler, B. L. Interspecies communication in bacteria. J Clin Invest 112, 1291–1299, https://doi.org/10.1172/JCI20195 (2003).
Antunes, L. C. et al. Bacteroides species produce Vibrio harveyi autoinducer 2-related molecules. Anaerobe 11, 295–301, https://doi.org/10.1016/j.anaerobe.2005.03.003 (2005).
Hsiao, A. et al. Members of the human gut microbiota involved in recovery from Vibrio cholerae infection. Nature 515, 423–426, https://doi.org/10.1038/nature13738 (2014).
Thompson, J. A., Oliveira, R. A., Djukovic, A., Ubeda, C. & Xavier, K. B. Manipulation of the quorum sensing signal AI-2 affects the antibiotic-treated gut microbiota. Cell Rep 10, 1861–1871, https://doi.org/10.1016/j.celrep.2015.02.049 (2015).
Collins, J. et al. Dietary trehalose enhances virulence of epidemic Clostridium difficile. Nature 553, 291–294, https://doi.org/10.1038/nature25178 (2018).
Tanghe, A., Van Dijck, P. & Thevelein, J. M. Determinants of freeze tolerance in microorganisms, physiological importance, and biotechnological applications. Adv Appl Microbiol 53, 129–176 (2003).
Zhu, Z. et al. Time course analysis of Candida albicans metabolites during biofilm development. J Proteome Res 12, 2375–2385, https://doi.org/10.1021/pr300447k (2013).
Bassler, B. L., Wright, M., Showalter, R. E. & Silverman, M. R. Intercellular signalling in Vibrio harveyi: sequence and function of genes regulating expression of luminescence. Mol Microbiol 9, 773–786 (1993).
Bushmanova E. A. D., Lapidus, A. & Przhibelskiy, A. D. rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. BioRxiv, https://doi.org/10.1101/420208 (2018).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Quinlan, A. R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr Protoc Bioinformatics 47, 11 12 11–34, https://doi.org/10.1002/0471250953.bi1112s47 (2014).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19, 455–477, https://doi.org/10.1089/cmb.2012.0021 (2012).
Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069, https://doi.org/10.1093/bioinformatics/btu153 (2014).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595, https://doi.org/10.1093/bioinformatics/btp698 (2010).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000).
Kanehisa, M., Sato, Y. & Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J Mol Biol 428, 726–731, https://doi.org/10.1016/j.jmb.2015.11.006 (2016).
Babicki, S. et al. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res 44, W147–153, https://doi.org/10.1093/nar/gkw419 (2016).
Fimlaid, K. A., Jensen, O., Donnelly, M. L., Siegrist, M. S. & Shen, A. Regulation of Clostridium difficile Spore Formation by the SpoIIQ and SpoIIIA Proteins. PLoS Genet 11, e1005562, https://doi.org/10.1371/journal.pgen.1005562 (2015).
Acknowledgements
We thank Gianfranco Donelli, and Claudia Vuotto, Microbial Biofilm Laboratory, IRCCS Fondazione Santa Lucia, Italy for providing the Bacteroides fragilis isolate and Prof Paul Williams, University of Nottingham for providing the Vibrio harveyi strain BB170. This was in part supported by a Seed grant from the Wellcome Warwick Quantitative Biomedicine Programme (Institutional Strategic Support Fund: 105627/Z/14/Z). We thank the Biotechnology and Biological Sciences Research Council (BBSRC) funded Midland Integrative Biosciences Training Partnership (MIBTP) studentships to LF and SR.
Author information
Authors and Affiliations
Contributions
R.S., L.F., S.J. designed and performed experiments for this study. A.M. and R.S. performed sequencing data analysis. R.S. and M.U. wrote the main manuscript and all authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Slater, R.T., Frost, L.R., Jossi, S.E. et al. Clostridioides difficile LuxS mediates inter-bacterial interactions within biofilms. Sci Rep 9, 9903 (2019). https://doi.org/10.1038/s41598-019-46143-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-46143-6
This article is cited by
-
Regulatory and innovative mechanisms of bacterial quorum sensing–mediated pathogenicity: a review
Environmental Monitoring and Assessment (2023)
-
Extracellular DNA, cell surface proteins and c-di-GMP promote biofilm formation in Clostridioides difficile
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
