
Abstract
Parasitic plants are known to discard photosynthesis thus leading to the deletion or loss of the plastid genes. Despite plastid genome reduction in non-photosynthetic plants, some nucleus-encoded proteins are transported back to the plastid to carry out specific functions. In this work, we study such proteins in Rafflesia cantleyi, a member of the holoparasitic genus well-known for producing the largest single flower in the world. Our analyses of three transcriptome datasets, two holoparasites (R. cantleyi and Phelipanche aegyptiaca) and one photosynthetic plant (Arabidopsis thaliana), suggest that holoparasites, such as R. cantleyi, retain some common plastid associated processes such as biosynthesis of amino acids and lipids, but are missing photosynthesis components that can be extensions of these pathways. The reconstruction of two selected biosynthetic pathways involving plastids correlates the trend of plastid retention to pathway complexity - transcriptome evidence for R. cantleyi suggests alternate mechanisms in regulating the plastidial heme and terpenoid backbone biosynthesis pathways. The evolution to holoparasitism from autotrophy trends towards devolving the plastid genes to the nuclear genome despite the functional sites remaining in the plastid, or maintaining non-photosynthetic processes in the plastid, before the eventual loss of the plastid and any site dependent functions.
Similar content being viewed by others


Introduction
Chloroplasts have long been recognized for their role in supporting plant growth and development1. Generally, chloroplast-localized proteins are either responsible for the localization and transport functions within the chloroplast or for chloroplast translation1 to carry out biosynthesis of fatty acids, amino acids, vitamins and nucleotides. Plastidial activities have also been shown to be involved in the synthesis of several plant hormones such as abscisic acids that are derived from isoprenoids, gibberrelins and brassinosteroids2. Non-photosynthetic parasitic plants source their carbon requirements directly from their respective hosts and the resulting relaxed selective pressures can be seen on their much reduced plastid genome3. As holoparasites adopt heterotrophy, many genes required for their original functions are believed to have been transferred to the nucleus4 or are lost. This phenomenon is also observed for other plants where the chloroplast genes are transferred to the nuclear genome as a result of evolutionary progression5. These genes are encoded in the nucleus, translated in the cytoplasm and their products are then transported back to the plastid5,6. The remnant plastid structure provides an overview of how a plastid genome evolves and its residual functions reveal the possible functionality of a reduced plastome. However, a recent study suggests that the chloroplast genome in the genus Rafflesia may be absent or present at very low levels, where plastidial fragments are only detected in sizes ranging from 104 to 1,026 bp7. Likewise, work on the non-photosynthetic green alga genus, Polytomella, revealed that they harbour plastids without a genome8.
The habitat of the rafflesiaceae family that the Rafflesia genus belongs to are confined to the tropical rainforests of Southeast Asia9. The species used in this work, Rafflesia cantleyi, is an obligate holoparasite that depends entirely on its host plant, Tetrastigma rafflesiae, for all nutrients including inorganic matter such as water10. In a comparative study of parasitic plants, Phelipanche aegyptiaca, a holoparasite belonging to the Orobanchaceae family was reported to maintain a complete, expressed and partially pressurized chlorophyll synthesis pathway despite its heterotrophic nature11. In parasites where the photosynthetic apparatus has been abandoned; the loss of selection pressure on the plastid genome, or more critically on the photosynthesis-related genes has been reported12. In reality, plastidial functions rely on both plastid- and nucleus-encoded proteins13. The genes involved in the plastid translational apparatus are also confined to both the plastid and the nucleus divisions14. Since most plastid genomes are functional, including in holoparasitic plants, nucleus-encoded proteins targeted to the plastid that compensate for gene losses are of great interest in understanding the residual functions of the reduced plastid in holoparasites.
Convolvulaceae and Orobanchaceae are two families that are often used for studying the evolution of holoparasitism from hemiparasitism because they have species ranging from hemi- to holoparasites15. We report here the results of a comparative survey of nucleus-encoded proteins targeting the plastid using RNA-Seq data obtained from Lee et al.16 and public databases. We observed that R. cantleyi possesses a relatively versatile nucleus-encoded plastid (NEP) RNA polymerase translation that conserves some common plastidial functions while adopting alternative route(s) to compensate for gene loss. The readily accessible bud transcriptome data of a holoparasite, P. aegyptiaca, along with the photosynthetic model plant Arabidopsis thaliana were used for comparison against our R. cantleyi subject.
Results
Genome assembly and annotation of the organellar genes
Roche 454 shotgun sequencing of the R. cantleyi genome generated a total of 1,091,861 high quality reads with an average length of 289 bp. Following sequence clustering, 607,596 DNA sequences were obtained and assembled into 4,944 contigs and 302,176 singletons. Classification and annotation of the 4,944 contigs using blastx and Blast2GO revealed a prevalence of mitochondrial genes. The singletons were therefore used instead for gene detection. Following BLAST annotation, the singletons were parsed to Blast2GO resulting in 1,799 out of 302,176 singletons being functionally annotated. Of these 1799 functionally annotated singletons, 217 were identified to be of plastid-origin, and 16 sequences were confined to homologs localized in the thylakoid (Fig. 1). Since many, if not most, non-photosynthetic organisms retain a functional plastid despite losing their photosynthetic genes, we focused the subsequent analyses on the nucleus-encoded plastid-targeting proteins commonly known to be present in photosynthetic plants that are responsible for several biosynthetic processes other than photosynthesis13. These subsequent analyses were carried out using transcriptome data.

Genome sequence distribution of annotated singletons of Rafflesia cantleyi according to cellular location by Blast2GO. A total of 1799 singletons were annotated and categorized according to their cellular location. From that, 217 sequences were found to be plastid-localized, out of which 16 are of thylakoid origin. This chart does not show the abundance of nucleus-encoded plastid-targeting genes, thus implying more genes are involved in plastid metabolic processes.
Identification and functional implication of nucleus-encoded plastid-targeting proteins
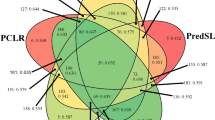
A total of three transcriptome data sets were compared in this study. The R. cantleyi transcriptome was sequenced in-house, producing 40,768,187 high quality reads16. The consensus contigs of P. aegyptiaca17 and the raw transcriptome data for A. thaliana were obtained online as described in the methodology section. After transcriptome assembly, the numbers of transcripts produced were 18,053 for R. cantleyi and 43,783 for A. thaliana, whereas 68,683 contigs were available for P. aegyptiaca. Putative nucleus-encoded proteins with the presence of a transit peptide detected for each data set were 150 for R. cantleyi, 198 for P. aegyptiaca and 272 for A. thaliana (Fig. 2a). The full putative protein list is given in the same order as Supplementary Dataset S1a, S1b, S1c. On average, the lengths of predicted transit peptides ranged from 10 to 80 bp. Since each of the transcriptome sizes differed, the lists of putative plastid-localized proteins could perhaps better serve as a reference for future experimental protein characterization rather than as a definitive means of revealing the true functions of the retained plastid of R. cantleyi. To increase the confidence level that these NEPs were putatively plastid localized, subcellular localization analysis was performed. This resulted in a further reduction of the plastid-localized proteins to 10 for R. cantleyi, 19 for P. aegyptiaca and 57 for A. thaliana (Fig. 2b). The full putative protein list is given in the same order as Supplementary Dataset S2a, S2b, S2c. The predicted nucleus-encoded plastid-targeting proteins in R. cantleyi and other plants do not represent the complete set of plastid-targeting proteins available for each plant. For this work, we focused on those with relevance to genes contained within the GreenCut2 Resource13 along with a predicted chloroplast transit peptide and a predicted localization to the plastid.

Identification of nucleus-encoded plastid-targeting proteins across Rafflesia cantleyi, Phelipanche aegyptiaca and Arabidopsis thaliana using the criteria described in methods. Putative nucleus-encoded plastid-targeting proteins with a predicted transit peptide (a); a predicted transit peptide and are targeted to the chloroplast (b) were sorted according to the GreenCut2 functional classification system. Nucleus-encoded plastid-targeting proteins retrieved from both non-photosynthetic holoparasites and photosynthetic green plants are presented.
Next, the putative plastid-localized proteins were compared based on the MapMan annotation provided in the GreenCut2 inventory. Of the 14 GO annotation groups; “protein”, “transport”, “nucleic” activities and “other” processes that consisted of RNA regulation, fibrillins, amino acid and minor carbohydrate metabolism made up the four dominant functional categories in the three species surveyed. Photosynthetic genes were mostly observed for A. thaliana and some for P. aegyptiaca, whereas the Rafflesia transcriptome did not show any significant presence of such genes. Functional categories of “carbohydrate”, “cell cycle” and “cofactors” were represented by fewer numbers of nucleus-encoded plastid-targeting proteins, possibly due to the classification parameters where some were instead grouped under “protein” and “other” categories. In this study, genes that were observed to be more commonly expressed than others (Table 1), were considered as components of well-conserved regulatory pathways not only for green photosynthetic plants but also for non-photosynthetic parasites. Some pathways and regulatory steps such as transfer of activated nucleotide sugars to acceptor molecules by glycosyltransferase18; modulation of the redox environment via the reduction of disulfide bridges in enzymes by thioredoxin19; mediation of TCA, glyoxylate bypass, amino acid synthesis, exchange of metabolites and gluconeogenesis by malate dehydrogenase20; and detoxification-associated pathways by glutathione s-transferase21 were deemed to remain functional except those specific for food production. In addition, the location of each gene as annotated may be indicative of the cellular location where they carry out their functions.
The transcription status of both NEP and non-NEP related genes were annotated based on their presence in the available transcriptome data. In this study, riboflavin synthase and lysophosphatidic acid acyltransferase, known for participating in riboflavin metabolism, and phosphatidic acid biosynthesis, were investigated using a phylogenetic approach (Supplementary Fig. S1). These genes were chosen due to their importance in providing indispensable precursors or intermediates for the plant’s survival. The shorter distance between Rafflesia and its known close relatives suggests that these genes were unlikely acquired from its host through horizontal gene transfer but were transcribed and expressed to fulfill specific biological roles. For instance, riboflavin synthase serves to produce vitamin B2, a precursor that further supplies essential cofactors for several cellular processes such as the citric acid cycle, fatty acid oxidation, and mitochondrial electron transport, therefore showing the importance of riboflavin accessibility22.
Pathway reconstruction in R. cantleyi
Two pathways were reconstructed to contrast the differences between a photosynthetic and a non-photosynthetic plant. These two pathways were chosen based on their retained presence in both parasitic and non-parasitic plants. The first pathway is the porphyrin biosynthesis pathway. Intermediate products of heme biosynthesis were observed for some genes but this pathway appears to end at heme production in R. cantleyi (Fig. 3). As expected, chlorophyll synthesis genes were not transcribed in R. cantleyi. Although identical initial steps for heme production and chlorophyll synthesis were present, the expression of genes directly involved in chlorophyll synthesis were not observed, thus alluding that this process no longer takes place in this holoparasite.

Analysis of heme biosynthesis pathway using genome and transcriptome data. This pathway consists of genes that mapped to that of Arabidopsis thaliana col heme biosynthesis I embedded within Plant Metabolic Network database. At the genome level, all ten genes were present whereas only eight genes (labeled with red box) were detected at transcript level for Rafflesia cantleyi. Using our assembled Arabidopsis transcriptome, all ten genes required in this process were retrieved.
Reconstruction of the terpenoid backbone biosynthesis pathway (Fig. 4) enabled the detection of genes that are involved in the mevalonate pathway. R. cantleyi was observed to be similar to other heterotrophic plants where the cytoplasm becomes the site for this biosynthetic process. This suggests that the parasite potentially takes up an alternative path within the cytoplasm due to the reduction of the plastid genome.

Reconstructed terpenoid backbone biosynthesis for Rafflesia cantleyi at transcript level. This pathway was reconstructed based on the detection of transcripts present. Of the (a) two mechanisms present for IPP production, Rafflesia is found to consist of only the (b) genes present in cytoplasm but not (c) genes in the plastid. Rafflesia-containing enzymes with EC numbers include: 2.3.1.9, acetyl-CoA c-acetyltransferase; 2.3.3.10, hydroxymethylglutaryl-CoA synthase; 2.7.1.36, mevalonate kinase; 2.7.4.2, phosphomevalonate kinase; 4.1.1.33, diphosphomevalonate decarboxylase; 2.5.1.1, 2.5.1.10, farnesyl diphosphate synthase; 2.5.1.92, (2z,6z)-farnesyl diphosphate synthase; whereas enzymes confined to the plastid were labeled with EC numbers: 2.2.1.7, 1-deoxy-D-xylulose-5-phosphate synthase; 1.1.1.267, 1-deoxy-D-xylulose-5-phosphate reductoisomerase; 2.7.7.60, 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase; 2.7.1.148, 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol kinase; 4.6.1.12, 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase; 1.17.7.1, (E)-4-hydroxy-3-methylbut-2-enyl-diphosphate synthase; 1.17.7.3, (E)-4-hydroxy-3-methylbut-2-enyl-diphosphate synthase; 1.17.7.4, 4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase; 5.3.3.2, isopentenyl-diphosphate Δ-isomerase; 4.2.3.27, isoprene synthase.
Analysis of Rafflesia cantleyi orthologous NEP genes
The ten putative nucleus-encoded plastid-targeting proteins (Supplementary Dataset S2a) of Rafflesia were used to search for their orthologs using OrthoMCL23. The orthologous groups identified for eight putative proteins are listed in Table 2. The orthologous groups are made up of biosynthetic and metabolic processes, regulation of transcription and response to stress. This characterization according to GO biological processes and molecular function reveals the potentially retained functions in the Rafflesia plastid.
Discussion
In this work, we compared the differences in detectable transcripts of nuclear genes encoding for plastid proteins in Rafflesia, Phelipanche and Arabidopsis. Variations in expression abundance plotted for each species reveals a relatively consistent trend of some housekeeping regulatory pathways for all species investigated, including the parasitic ones. Plastids are capable of transforming from one type to another, accompanied by the alteration of plastid proteome composition24. Despite some differences, both tissue and environmental signals induce changes in the plastid proteomes24. Therefore, a core set of essential genes is expected to be transcribed in each species. After the adoption of parasitism, relaxed selective constraints may have resulted in further plastome reduction. Additionally, the continued nonfunctionality of the plastome in parasitic plants contributes to the accumulation of mutagenic factors including microsatellites, long homopolymer tracts, forward or palindromic repeats, and a poor GC content25. However, these aspects of plastome evolution are not discussed here. We instead focus on how some nucleus encoded plastid genes may have been repurposed or have remained functional because their products are also core components of other non-photosynthesis pathways.
As most ndh and chlororespiratory genes are lost, the preferably retained ribosomal proteins and tRNAs in parasites may indicate an existence of a functional plastid DNA shift to the nuclear genomes25. In general, the loss of a subset of nonfunctional-related ribosomal proteins leads to a decline in plastidial translational capacity and photosynthetic performance26. Moreover, a complete loss of the plastidial and nuclear genes involved in the plastid DNA expression system possibly indicates the absence of a plastid genome, as proposed for the Polytomella genus8. In the case of Rafflesia, several plastidial ribosomal proteins along with the nucleus-encoded plastid-targeting genes were detected from the transcript data. Identification and characterization of the nucleus-encoded plastid-targeting proteins may therefore unveil the possible remaining plastidial functions. In addition, these studies provide a comparative view of the plastid genome evolution between R. cantleyi and other representatives under relaxed selective constraints.
In this study, the engagement of transit peptides and subcellular localization in the identification of nucleus-encoded plastid-targeting proteins helps to eliminate possible false positives (Fig. 2a,b), therefore, this analysis was not aimed at elucidating the true number of plastid-localized proteins. We had used the GreenCut2 inventory that was derived from protein data to identify the set of plastid-localized proteins in different species. Despite the differences between lineages, transit peptides possess, there are some similarities in amino acid composition such as the relatively high abundance of serine and threonine residues and that these residues are positively charged. This allows for transit peptide prediction through different software. We have used only the transcriptome from the flower to give an overview of the retained plastid function at the final stage of the parasites’ life cycle. Despite the true number of >2,500 plastid-localized proteins reported for flowering plants, these numbers vary greatly across species. Factors such as the sample developmental stage, pipeline used and the filtering criteria also contribute to the differences in numbers. The prediction of transit peptides and subcellular localization improved and increased the confidence level of the final output. We had also filtered the candidates through bioinformatic prediction with a cutoff ≥0.500 for TargetP analysis and this is further supplemented with manual inspection of the results. These steps help to reduce false-positives. This approach may most likely underestimate the true number of plastid-localized genes but serves to achieve the objective of identifying such genes for further analysis. We must there make clear that these results are by no means an exhaustive or complete prediction regarding the true extent of plastid localized genes. Further identification of the sequence motifs were not carried out due to the presence of multiple but non-conserved groups of nucleobases present in different transit peptides27.
The reconstruction of two selected pathways revealed their distinct mechanisms in R. cantleyi. The synthesis of heme is carried out by both prokaryotes and eukaryotes due to it being necessary for the function of cytochromes, chlorophylls, phycobilins, and the corrin nucleus of vitamin B1228. In non-photosynthetic organisms, heme participates in the transfer of electrons and the binding of diatomic gases29. The incomplete set of genes expressed in Rafflesia during heme biosynthesis has raised questions as to whether the plant achieves equally effective heme production. In comparison, the premature bud of the photosynthetic Arabidopsis seems to contain a complete set of expressed heme biosynthetic genes. It is known that even though heme biosynthesis mainly takes place in the cytoplasm and mitochondrion, synthesis of δ-amino-laevulinic acid (ALA), a key precursor, occurs in the plastid30. Previous studies have shown that non-photosynthetic plants retain the trnE gene and their associated specific transcription machinery in order to equip heme biosynthesis with mitochondrial cytochromes, P450 cytochromes and other necessary oxidative enzymes31. This is congruent with the proposed trnE hypothesis, claiming that the retention of a non-photosynthetic plastid is responsible for heme biosynthesis30. Heme is considered to be indispensable and is synthesized by all cells32. From the available data, we postulate that Rafflesia achieves its heme production in the plastid but discards some later stages of tetrapyrrole synthesis such as chlorophyll and perhaps, phytochromobilin formation.
Due to the differences in feeding nature, the terpenoid backbone biosynthesis between parasitic and non-parasitic plants is expected to exhibit two different mechanisms. Generally, most eukaryotes, archaebacteria, fungi, the cytosol and mitochondria of plants use the mevalonate (MVA) pathway33. This phenomenon is different in most eubacteria where the non-mevalonate (MEP) pathway appears to be the only pathway deployed for the biosynthesis of their isoprenoid precursors34. Plastid-bearing organisms, including plants, apicomplexan parasites such as Plasmodium falciparum (malaria) and Toxoplasma gondii (toxoplasmosis), also adopt the MEP pathway35. One intermediate product of terpenoid backbone biosynthesis produced from both the MVA and MEP pathways is IPP, and all isoprenoids are derived from this five-carbon (C5) compound along with its isomer dimethylallyl diphosphate (DMAPP)34,36. The starting substrates for both the MVA and MEP pathways are inherited from glycolysis, which are present in both photosynthetic and nonphotosynthetic plants (Fig. 4). The choice of which pathway to enter may possibly be affected by the site of synthesis; be it cytoplasmic- or plastid-located, or whether an active transport process occurs between the two organelles37. The synthesis of aromatic compounds in the cytoplasm suggests that the plastid in Rafflesia has been reduced or pseudogenized of this alternative route in providing IPP. The detection of the plastidial remnants at genome level, but not during transcriptome level analysis, has led to this interpretation. Intermediate substrates needed for sterols, sesquiterpenes and the side chain of ubiquinone biosynthesis are therefore provided by cytosolic metabolism38. The second hypothesis regarding this phenomenon is the lack of the above secondary metabolite biosynthesis in the parasitic R. cantleyi. The partial or selective reduction of this biosynthesis pathway can also be illustrated by the lack of detectable gene sequences participating in the carotenoid pathway. Conversely, the carotenoid pathway is present in the Polytomella genus but found to be at various stages of degradation in the four species studied39.
Ideally, the abundance of nuclear plastid DNA-like sequences (NUPTs) in nuclear genomes correlates well with the number of plastids present in an organism40. This phenomenon is found to be consistent across terrestrial plants, green algae, apicomplexans and stramenopiles. However, evolutionary parasitism has propelled plastid reduction and the extent of their viability depends on the degree of parasitism and plastid metabolic complexity41. The more diverse plastid of Phelipanche compared to Rafflesia may indicate a more recent autotrophic ancestor. Orobanchaceae was reported to have undergone massive gene loss and pseudogenisation42. If this was the case for Orobanchaceae, a greater gene loss in Rafflesiaceae would not be surprising. This can be attributed to the non-photosynthetic status of all Rafflesia species whereas some species in Orobanchaceae still retain either a complete or a collectively constrained chlorophyll synthesis pathway11. In contrast, non-photosynthetic tissues of photosynthetic plants are found to contain a more complete set of genes even though they do not require an active photosynthetic apparatus at this stage. Studies on the plastidial proteins encoded by the nucleus provide a rudimentary view of the possibly retained functions known to be taking place in plastids. In addition, retention of NEP-related pathways in plastid indicates an inter-connection among the actual pathways sited in the plastids to the nuclear genome encoding the genes41.
With the currently available technology and resources, it is highly unlikely that a large scale comparative experiment can be executed to focus solely on the question of the loss of photosynthetic capacity in diverse plants from different geographical regions. Furthermore, the cultivation and tissue culture of parasitic plants are extremely difficult thus highly dependent on the availability of field samples. However, the availability of publicly accessible genome and transcriptome data can be analysed and certain contextual insights can be extracted from them. Such approaches undoubtedly have limitations and care must also be made in extrapolating functional information from such datasets. Nevertheless, they can prove to be useful starting points for further investigations as demonstrated in this work.
Conclusions
Comparative studies of nucleus-encoded plastid-targeting proteins of non-photosynthetic parasitic plants and the photosynthetic ones reveal the transcription status of these proteins and some prominent processes pertaining to the plastid despite plastid reduction. Fundamental roles such as amino acid biosynthesis and nucleic acid metabolism are two such examples that would seldom be discarded by any plants. The phylogeny of Rafflesia for two selected nucleus-encoded proteins showed no evidence that the genes encoding those functions were acquired by horizontal gene transfer, implying that they are expressed by the parasitic plant itself to fulfill its biological roles. The available evidence however suggests that the plastid genome may devolve to the nuclear genome and necessary functions are transported back to the plastid or the residual encoded plastidial functions partake in essential processes that are not related to phtosynthesis.
Methods
DNA sequencing and genome sequence annotation
The Rafflesia cantleyi flower sample was collected from Pahang, Malaysia. DNA was extracted from the perigone lobe tissue of the Rafflesia flower using a modified CTAB protocol43. Extracted genomic DNA was checked for quantity and quality using the NanoDrop 2000c machine and conventional gel electrophoresis. Ligation of specific adaptors and emulsion PCR amplification were carried out following the manufacturer’s protocol. After quality assessment, the sample was run on the Roche GS FLX Titanum sequencer. Shotgun sequencing reads generated were filtered of adaptors, and short (<50 bp) and low quality (QPhred < 20) sequences using perl scripts. Redundant reads were removed using cd-hit44 prior to genome sequence assembly using Newbler45 with default parameters (minimum overlap identity 90 bp; minimum overlap length 40 bp). All contigs and singletons were queried for gene information using BLAST v2.2.26 (http://www.ncbi.nlm.nih.gov/) (BLASTX, 1e-6)46 and Plant Metabolic Network (http://www.plantcyc.org/) (BLASTX, 1e-10)47. In addition, Blast2GO48 was used to annotate and classify genes according to their cellular distribution, molecular function and biological process.
RNA sequence assembly and annotation
Total RNA of Rafflesia flower was isolated from the same sample using a method by López-Gomez et al.49. RNA integrity was assessed using Agilent BioAnalyzer (Agilent Technologies, USA) and the quality was examined using Spectrophotometer ND-1000 (NanoDrop, USA). After cDNA preparation and library construction using TruSeq RNA Sample Prep kit, the sample was sequenced using the Illumina Solexa platform16. The raw data was preprocessed and annotated using BLAST v2.2.2646, and Blast2GO48. SRA archive file for Arabidopsis bud (#SRR544881) tissues was downloaded from the NCBI Sequence Read Archive (http://www.ncbi.nlm.nih.gov/sra). Transcript sequences for Phelipanche aegyptiaca bud were downloaded from the Parasitic Plant Genome Project (http://ppgp.huck.psu.edu/download.php)17. All files, except for contigs downloaded for P. aegyptiaca, were trimmed for short (<50 bp) and low quality (QPhred < 20) sequences using Perl scripts. Sequences were then checked for the presence of sequencing primers using FastQC v0.7.050. Clean and high quality reads were then subjected to sequence assembly using VelvetOptimiser (https://github.com/Victorian-Bioinformatics-Consortium/VelvetOptimiser)51 and Velvet51 to obtain an optimised k-mer. Oases52 was then used to produce flower transcripts from Velvet contigs. Assembly for the pre-processed Arabidopsis sequences was performed using Trinity53 and Oases52. All transcript sets were then queried using BLAST v2.2.26 (BLASTX, 1e−5)46.
Identification of nucleus-encoded plastid-targeting proteins
For each transcriptome dataset, the annotated transcripts were compared with the GreenCut2 Inventory13 to produce a list of candidates for nucleus-encoded plastid-targeting proteins. The six-frame translation of the candidate plastid-localized genes was performed using EMBOSS Transeq (http://ww.ebi.ac.uk/Tools/st/emboss_transeq/). Coding sequences were predicted from the transcripts using Transdecoder54 on Galaxy v1.0.3.055. The corresponding translated peptide sequences from EMBOSS Transeq were subjected to prediction for the presence of a plastid-targeting transit peptide using ChloroP (http://www.cbs.dtu.dk/services/ChloroP/)56. Initial lists of the candidate plastid-localized proteins with a predicted transit peptide were checked for their corresponding coding sequences. Candidate plastid-localized genes that lacked a predicted coding sequence were discarded from the lists. Next, the predicted peptide sequences of the candidates were used for subcellular localization prediction using TargetP v1.1 (http://www.cbs.dtu.dk/services/TargetP/)57 with cutoff ≥0.500. A list consisting of the final putative plastid-localized proteins was compiled from the prediction output for each dataset. The lists of putative plastid-localized proteins after ChloroP and TargetP prediction were categorized into their functional groups according to the classification system in the GreenCut2 Inventory. For two selected plastid-localized proteins, RAxML (http://embnet.vital-it.ch/raxml-bb/)58 was used for phylogenetic tree construction. The putative nucleus-encoded plastid-targeting proteins identified for R. cantleyi were searched for orthologous genes using OrthoMCL v5 (http://orthomcl.org/orthomcl/)23.
Pathway mapping and reconstruction
Two plastid-regulating pathways commonly retained in both photosynthetic and non-photosynthetic plants were selected for Rafflesia pathway reconstruction. Transcript data was used for gene detection and pathway mapping. Briefly, transcript IDs in GenBank format were extracted from each blast output file and were converted to uniprotAC and KO format using an embedded tool in UniPROT (http://www/uniprot.org/)59. To get a first glimpse of the expressed transcripts and their respective pathways, KO IDs were mapped against KEGG reference (http://www.genome.ad.jp/kegg/) pathways. The reference pathways selected for pathway reconstruction were downloaded from KEGG (http://www.genome.ad.jp/kegg/)60. The detected transcripts for each regulatory step were manually inspected and included in the newly reconstructed pathways.
References
Bryant, N., Lloyd, J., Sweeney, C., Myouga, F. & Meinke, D. Identification of nuclear genes encoding chloroplast-localized proteins required for embryo development in Arabidopsis. Plant Physiol. 155, 1678–1689 (2011).
Lopez-Juez, E. & Pyke, K. A. Plastids unleashed: their development and their integration in plant development. Int. J. Dev. Biol. 49, 557–577 (2005).
Morden, C. W., Wolfe, K. H., dePamphilis, C. W. & Palmer, J. D. Plastid translation and transcription genes in a non-photosynthetic plant: intact, missing and pseudo genes. Embo. J. 10, 3281–3288 (1991).
Gile, G. H. & Keeling, P. J. Nucleus-encoded periplastid-targeted EFL in chlorarachniophytes. Mol. Biol. Evol. 25, 1967–1977 (2008).
Schein, A. I., Kissinger, J. C. & Ungar, L. H. Chloroplast transit peptide prediction: a peek inside the black box. Nucleic Acids Res. 29, E82 (2001).
de Koning, A. P. & Keeling, P. J. Nucleus-encoded genes for plastid-targeted proteins in Helicosporidium: functional diversity of a cryptic plastid in a parasitic alga. Eukaryot. Cell 3, 1198–1205 (2004).
Molina, J. et al. Possible loss of the chloroplast genome in the parasitic flowering plant Rafflesia lagascae (Rafflesiaceae). Mol. Biol. Evol. 31, 793–803 (2014).
Smith, D. R. & Lee, R. W. A plastid without a genome: evidence from the nonphotosynthetic green algal genus Polytomella. Plant Physiol. 164, 1812–1819 (2014).
Bendiksby, M. et al. Elucidating the evolutionary history of the Southeast Asian, holoparasitic, giant-flowered Rafflesiaceae: pliocene vicariance, morphological convergence and character displacement. Mol. Phylogenet. Evol. 57, 620–633 (2010).
Nais, J. Rafflesia of the world. Natural History Publications, Kota Kinabalu, Malaysia (2001).
Wickett, N. J. et al. Transcriptomes of the parasitic plant family Orobanchaceae reveal surprising conservation of chlorophyll synthesis. Curr. Biol. 21, 2098–2104 (2011).
Krause, K. From chloroplasts to “cryptic” plastids: evolution of plastid genomes in parasitic plants. Curr. Genet. 54, 111–121 (2008).
Karpowicz, S. J., Prochnik, S. E., Grossman, A. R. & Merchant, S. S. The GreenCut2 resource, a phylogenomically derived inventory of proteins specific to the plant lineage. J. Biol. Chem. 286, 21427–21439 (2011).
Yamaguchi, K. & Subramanian, A. R. The plastid ribosomal proteins. J. Biol. Chem. 275, 28466–28482 (2000).
Nickrent, D. L. Parasitic plants of the world. Parasitic Plants of the Iberian Peninsula and Balearic Islands 2, 7–27 (2002).
Lee, X. W. et al. Perigone lobe transcriptome analysis provides insights into Rafflesia cantleyi flower development. PLoS ONE 11, e0167958 (2016).
Yang, Z. et al. Comparative transcriptome analyses reveal core parasitism genes and suggest gene duplication and repurposing as sources of structural novelty. Molecular biology and evolution 343 (2014).
Kapitonov, D. & Yu, R. K. Conserved domains of glycosyltransferases. Glycobiol. 9, 961–978 (1999).
Chibani, K., Wingsle, G., Jacquot, J.-P., Gelhaye, E. & Rouhier, N. Comparative genomic study of the thioredoxin family in photosynthetic organisms with emphasis on Populus trichocarpa. Molecular Plant 2, 3080322 (2009).
Musrati, R. A., Kollárová, M., Mernik, N. & Mikulášová, D. Malate dehydrogenase: distribution, function and properties. Gen. Physiol. Biophys. 17, 193–210 (1998).
Dixon, D. P., Lapthorn, A. & Edwards, R. Plant glutathione transferases. Genome Biol. 3, reviews3004.3001–3004.3010 (2002).
Jordan, D. B., Bacot, K. O., Carlson, T. J., Kessel, M. & Viitanen, P. V. Plant riboflavin biosynthesis. J. Biol. Chem. 274, 22114–22121 (1999).
Li, L., Stoeckert, C. J. Jr. & Roos, D. S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003).
Liebers, M. et al. Regulatory shifts in plastid transcription play a key role in morphological conversions of plastids during plant development. Front. Plant Sci. 8 (2017).
Wicke, S. et al. Mechanisms of functional and physical genome reduction in photosynthetic and nonphotosynthetic parasitic plants of the broomrape family. The Plant Cell 25, 3711–3725 (2013).
Romani, I. et al. Versatile roles of Arabidopsis plastid ribosomal proteins in plant growth and development. Plant J. 72, 922–934 (2012).
Lee, D. W. et al. Arabidopsis nuclear-encoded plastid transit peptides contain multiple sequence subgroups with distinctive chloroplast-targeting sequence motifs. The Plant Cell 20, 1603–1622 (2008).
Oborník, M. & Green, B. R. Mosaic origin of the heme biosynthesis pathway in photosynthetic eukaryotes. Mol. Biol. Evol. 22, 2343–2353 (2005).
Kořený, L., Oborník, M. & Lukeš, J. Make it, take it, or leave it: Heme metabolism of parasites. PLOS Pathogens 9, e1003088 (2013).
Barbrook, A. C., Howe, C. J. & Purton, S. Why are plastid genomes retained in non-photosynthetic organisms? Trends Plant Sci. 11 (2006).
Howe, C. J. & Smith, A. Plants without chlorophyll. Nature 349 (1991).
Atteia, A., van Lis, R. & Beale, S. I. Enzymes of the heme biosynthetic pathway in the nonphotosynthetic alga Polytomella sp. Eukaryot. Cell 4, 2087–2097 (2005).
Goldstein, J. L. & Brown, M. S. Regulation of the mevalonate pathway. Nature 343, 425–430 (1990).
Rodríguez-Concepción, M. & Boronat, A. Elucidation of the methylerythritol phosphate pathway for isoprenoid biosynthesis in bacteria and plastids. A metabolic milestone achieved through genomics. Plant Physiol. 130, 1079–1089 (2002).
Odom, A. R. Five questions about non-mevalonate isoprenoid biosynthesis. PLoS Pathog. 7, e1002323 (2011).
Bick, J. A. & Lange, B. M. Metabolic cross talk between cytosolic and plastidial pathways of isoprenoid biosynthesis: unidirectional transport of intermediate across the chloroplast envelope membrane. Arc. Biochem. Biophys. 415, 146–154 (2003).
Rohmer, M. The discovery of a mevalonate-independent pathway for isoprenoid biosynthesis in bacteria, algae and higher plants. Nat. Prod. Rep. 16, 565–574 (1999).
Vranová, E., Coman, D. & Gruissem, W. Structure and dynamics of the isoprenoid pathway network. Mol. Plant 5, 318–333 (2012).
Asmail, S. R. & Smith, D. R. Retention, erosion, and loss of the carotenoid biosynthetic pathway in the nonphotosynthetic green algal genus Polytomella. New Phytol. 209, 899–903 (2016).
Smith, D. R., Crosby, K. & Lee, R. W. Correlation between nuclear plastid DNA abundance and plastid number supports the limited transfer window hypothesis. Genome Biol. Evol. 3, 365–371 (2011).
Borza, T., Popescu, C. & Lee, R. W. Multiple metabolic roles for the nonphotosynthetic plastid of the green alga Prototheca wickerhamii. Eukaryot. Cell 4, 253–261 (2005).
Wolfe, K. H., Morden, C. W. & Palmer, J. D. Function and evolution of a minimal plastid genome from a nonphotosynthetic parastic plant. Proc. Natl Acad. Sci. USA 89, 10648–10652 (1992).
Doyle, J. J. & Doyle, J. L. A rapid DNA isolation procedure for small quantities of fresh lead tissue. Pytochemical Bulletin 19, 11–15 (1987).
Li, W.-Z. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Margulies, M. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380 (2005).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J Mol Biol 215, 403–410 (1990).
Schläpfer, P. et al. Genome-Wide Prediction of Metabolic Enzymes, Pathways, and Gene Clusters in Plants. Plant Physiology 173, 2041–2059 (2017).
Götz, S. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Res 36, 3420–3435 (2008).
López-Gómez, R. & Gómez-Lim, M. A. A method of extracting intact RNA from fruits rich in polysaccharides using ripe mango mesocarp. Hort Science 27, 440–442 (1992).
FastQC: a quality control tool for high throughput sequence data, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (2010).
Zerbino, D. R. & Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18, 821–829 (2008).
Schulz, M. H., Zerbino, D. R., Vingron, M. & Birney, E. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28, 1086–1092 (2012).
Grabherr, M. G. et al. Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotechnol 29, 644–652 (2011).
Haas, B. J. et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc 8, 1494–1512 (2013).
Afgan, E. et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 46, 537–544 (2018).
Emanuelsson, O., Nielsen, H. & von Heijne, G. ChloroP, a neural network-based method for predicting chloroplast transit peptides and their cleavage sites. Protein Sci. 8, 978–984 (1999).
Emanuelsson, O., Nielsen, H., Brunak, S. & von Heijne, G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J Mol Biol 300, 1005–1016 (2000).
Stamatakis, A., Hoover, P. & Rougemont, J. A rapid bootstrap algorithm for the RAxML Web servers. Systematic biology 57, 758–771 (2008).
Consortium, T. U. Activities at the Universal Protein Resource (UniProt). Nucleic Acids Res. 42, D191–D198 (2014).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000).
Acknowledgements
The work was supported financially by Research University Grants from Universiti. Kebangsaan Malaysia (Grant No. UKM-AP-KPB-18-2010, LAUREATE-2013-001, GUP-2016-008 and DIP-2017-013).
Author information
Authors and Affiliations
Contributions
S.M.N., M.F.R. and K.L.W. conceived and designed the experiments. J.H.A. and M.A.A.J. collected the samples. S.M.N. performed the data analysis. X.W.L. carried out the flower transcriptome assembly. M.N.M.I. participated in the data generation and sequence assembly. R.M. contributed to the project design and manuscript revisions. S.M.N., M.F.R. and K.L.W. wrote the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ng, SM., Lee, XW., Mat-Isa, MN. et al. Comparative analysis of nucleus-encoded plastid-targeting proteins in Rafflesia cantleyi against photosynthetic and non-photosynthetic representatives reveals orthologous systems with potentially divergent functions. Sci Rep 8, 17258 (2018). https://doi.org/10.1038/s41598-018-35173-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-35173-1
Keywords
This article is cited by
-
The Sapria himalayana genome provides new insights into the lifestyle of endoparasitic plants
BMC Biology (2023)
-
Transcriptome analysis of Rafflesia cantleyi flower stages reveals insights into the regulation of senescence
Scientific Reports (2021)
-
The loss of photosynthesis pathway and genomic locations of the lost plastid genes in a holoparasitic plant Aeginetia indica
BMC Plant Biology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
