
Abstract
Papaya ringspot virus (PRSV) seriously limits papaya (Carica papaya L.) production in tropical and subtropical areas throughout the world. Coat protein (CP)- transgenic papaya lines resistant to PRSV isolates in the sequence-homology-dependent manner have been developed in the U.S.A. and Taiwan. A previous investigation revealed that genetic divergence among Hainan isolates of PRSV has allowed the virus to overcome the CP-mediated transgenic resistance. In this study, we designed a comprehensive RNAi strategy targeting the conserved domain of the PRSV CP gene to develop a broader-spectrum transgenic resistance to the Hainan PRSV isolates. We used an optimized particle-bombardment transformation system to produce RNAi-CP-transgenic papaya lines. Southern blot analysis and Droplet Digital PCR revealed that line 474 contained a single transgene insert. Challenging this line with different viruses (PRSV I, II and III subgroup) under greenhouse conditions validated the transgenic resistance of line 474 to the Hainan isolates. Northern blot analysis detected the siRNAs products in virus-free transgenic papaya tissue culture seedlings. The siRNAs also accumulated in PRSV infected transgenic papaya lines. Our results indicated that this transgenic papaya line has a useful application against PRSV in the major growing area of Hainan, China.
Similar content being viewed by others

Introduction
Papaya (Carica papaya L.) is a widely cultivated fruit crop in the tropics and sub-tropics. However, the production of papaya is seriously limited by papaya ringspot disease caused by Papaya ringspot virus (PRSV) worldwide. Infected papaya plants exhibit symptoms of yellowing, distortion, severe leaf mosaic, and the classic “ringspot” on fruit1. Viral infection impacts papaya growth, reduces fruit quality, and prevents fruit set2. It is in the genus Potyvirus and contains a monopartite, single-stranded, positive-sense RNA3. PRSV isolates are grouped into the papaya-infecting type (PRSV-P) that infects both papaya and cucurbits, and the cucurbit-infecting type (PRSV-W) that infects cucurbits but not papaya2,4. PRSV is mainly transmitted by aphids in a non-persistent manner5.
Papaya and Arabidopsis thaliana are members of the Brassicales. Genome sequence analysis revealed that the papaya has significantly fewer disease resistance genes than that of Arabidopsis. The papaya genome is significantly larger, at 372 mega base pairs (Mbp) vs. 145 Mbp6. However, there are fewer nucleotide-binding site (NBS)-containing R genes in papaya than in Arabidopsis, 54 vs. 174, respectively7. The natural source of lacking effective resistance makes the conventional breeding for resistance difficult7. Several methods are used to control papaya ringspot disease, including quarantine, geographic displacement, roguing, netting, cross-protection, and genetic modification of the host plant8. Because PRSV is rapidly and efficiently transmitted by aphids, the use of insecticides is impractical2. In Hawaii and Taiwan, a mild strain of PRSV, HA5-1, has been used to protect papaya plants against the infection by virulent strains of the virus9.
The first commercialized transgenic papaya carrying the PRSV CP gene was introduced to Hawaii in 1998 and saved the remains of the papaya industry10. However, CP-transgenic resistance of papaya is expressed in a nucleotide-sequence-homology-dependent manner11. Transgenic papaya cultivars have varying levels of resistance against PRSV isolates from other geographical regions. For example, isolates from the Bahamas, Florida and Mexico have delayed, mild symptoms. Isolates from Brazil and Thailand also have delayed symptoms, but the virus eventually overcomes their resistance12. The CP hemizygous line, ‘Rainbow,’ is also susceptible to PRSV isolates from Taiwan13. Resistance levels therefore depend on the variability among CP genes of the isolates8,14,15. We have previously reported that PRSV isolates from Hainan province, China, are highly variable16. The high levels of genetic divergence in PRSV isolates from Hainan is likely to be the cause of the failure of transgenic papaya lines that targets specific viral CP genes16.
The RNA-mediated resistance in papaya is based on post-transcriptional gene silencing (PTGS), a host defense response to foreign RNA11. PTGS-mediated transgenic resistance depends on the sequence homology between a transgene and the corresponding viral genome17. Transgenic papaya has been proven to have effective resistance to PRSV isolates from Hawaii, Taiwan, and other. In this work, we use the RNAi strategy to construct a transgene that targets the conserved region of the PRSV CP genes and confers broad-spectrum resistance to the diverse Hainan PRSV isolates.
Results
Vector construction and plant transformation
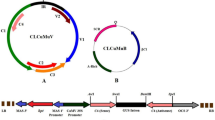
The genetic variability among PRSV isolates in Hainan was investigated in a previous study16. Sequence analysis of the 544-bp region of the Hainan PRSV isolates shared 97 to 100% identity (Supplementary Figure 1) were used to design RNAi hairpin structures. The constructed CP hairpin structure was inserted into pCAMBIA2300-35S-OCS to construct the plant expression RNAi vector (Fig. 1). The hairpin structure was validated by digestions with Sal I and Pst I (Supplementary Figure 2).

Components of the constructed RNAi vector pCAMBIA2300-35S-OCS. 35 S promoter indicated Cauliflower mosaic virus (CaMV) promoter. NOS terminator = nopaline synthase gene terminator, OCS terminator = octopine synthase terminator. NPTII = neomycin phosphotransferase gene. Sense and Anti-sense = conserved 544 bp fragment of CP gene and inverted repeat sequence.
Identification of transgenic lines by PCR
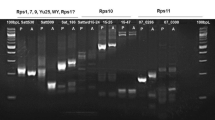
All plants regenerated from embryogenic callus under antibiotic selection were screened for transgenic events by PCR (Fig. 2). Primers amplifying a 760-bp fragment containing the 35 S promoter elements and complete CP hairpin region were used to verify presence of the target transgene (Table 1 and Fig. 2). Regenerated lines 242, 422, 474, and 537 were confirmed to be transgenic by PCR, while line 1280 was not transgenic.

PCR validation of the transgenic event in difference lines. CP1/CP2 primers amplified the 544-bp conserved region of the CP gene in sense orientation. 35S-F/CP2 primers were vector-specific, amplifying the 35 S promoter and conserved sense region of the CP gene. Papain F/ Papain R primers amplified the papaya reference gene. M = DNA molecular weight, P = plasmid DNA, SU = ‘SunUp’ DNA, SR = ‘SunRise’ DNA before transformation. The other numbers are candidate transgene lines. Arrows on the right panel indicated the expected PCR products. The full-length gels are presented in Supplementary Figure 3.
Southern blot analysis to confirm single insert in transgenic line
Digestion of line 474 genomic DNA with either Hind III or BamH I produced a Southern blot that showed a single band in each digest, indicating that there is only one insertion site in transgenic line 474 (Fig. 3).

Southern blot analysis confirmed a single insertion site in transgenic line 474. Plasmid and line 474 were digested with BamH I and Hind III separately, and the 544-bp CP gene conserved region was produced by PCR as a reference control. (A) Agarose gel image before transfer to nylon membrane. (B) Southern blot. The probe of CP fragment and DNA marker were labeled with Digoxigenin-11-dUTP individually, and mixed together before hybridization.
Using digital PCR to determine copy number in the transgenic lines
Analysis by ddPCR showed that the transgenic insertion in papaya line 474 contained 0.83 copy (about 1) of the CP gene. SunUp is homozygous for the CP gene at a single insertion site (CP/CP)18 and therefore has 2 copies of the transgene (Supplementary Figure 4).
hiTAIL-PCR used to locate the insertion site
The genomic location of the insert in transgenic line 474 was determined by hiTAIL-PCR (Fig. 4A). The result showed the insertion site was anchored on papaya chromosome VII, within supercontig_61 at base pair 717,141 (Carica papaya ASGPB v4.0, https://phytozome.jgi.doe.gov). The insertion did not lie within any predicted open reading frames (ORFs) (Fig. 4B).

High-efficiency thermal asymmetric interlaced PCR (hiTAIL-PCR) verified the insertion site. (A) Two rounds of hiTAIL-PCR for line 474. M = molecular marker; lane 1 = first round PCR; lane 2 = second round PCR; arrow indicates two products that were subsequently sequenced. (B) Structural diagram of the specific insertion site located in chromosome VII, supercontig 61 at 717141 bp. Primer 474-61 F/R were designed to amplify 192 bp in non-transgenic line. (C) Nested PCR with primers 474-61 F/61 R bracketing the insertion site and primers CP1/CP2 used to verify the insertion of the CP gene. Line 474 samples amplified as a PCR template produced two bands; one band (192 bp) was the same as the negative control for the insertion event, while the other band was about 1.2 kb.
Transformation-site-specific PCR to confirm the insertion
To further verify the authenticity of the insert, two primers, 474–61 F/474-61 R, were designed to amplify a 192-bp product containing and centered approximately on the insertion site (Fig. 4C). The site-specific primers produced one band of the expected size (192 bp), when amplifying genomic DNA of a non-transgenic control line, but no band was produced when genomic DNA of line 474 was used as template, due to the large size of the inserted sequence. Furthermore, nested PCR containing both a site-specific primer and CP reverse primers was designed to confirm the insertion site. PCR products from line 474 contain a 1.2-kb band (CP inserts + 35 S promoter + plasmid right border + plant genome right wing). Additionally, we also confirmed that line 474 was hemizygous (CP/-), since the nested primers produced both the 1.2-kb site-specific band and the 192-bp band of the unaltered genome (Fig. 4C).
Bioassays for resistance to Hainan PRSV isolates in transgenic line 474
Transgenic line 474 and a non-transgenic line 1280 were challenged with severe PRSV isolates of Hainan subgroups I, II and III, as well as a mixture of all three. The accumulation of viruses in inoculated plants was monitored every 4 days from 0 to 28 days after inoculation (DAI) (Fig. 5). Typical PRSV symptoms were observed in line 1280 at 12 DAI, and severe symptoms were observed at 24 DAI. In contrast, line 474 had no visual symptoms (Fig. 5A). Reverse transcription PCR (RT-PCR) using primers specific for the HC-Pro gene of PRSV was employed to semi-quantitatively measure PRSV replication in inoculated papaya plants. PRSV accumulation increased dramatically in line 1280 by 12 DAI, but there was no evidence of PRSV in line 474 over the course of the experiment (Fig. 5).

Bioassays of transgenic line 474 to determine resistance response to inoculation with a mixture of PRSV isolates F61d, F10d and F21d. (A) Plant inoculation bioassay. Typical PRSV symptoms were observed in non-transgenic line 1280 at 24 DAIs, while transgenic line 474 had no visual symptoms. IL indicates the inoculated leaf, DL indicates the detected leaf. (B) Reverse-transcription PCR of the HC-Pro gene (484 bp) was used to detect PRSV. Papain was used as internal reference gene (198 bp). PRSV accumulation in the transgenic and non-transgenic papaya was monitored 0, 4, 8, 12, 16, 20, 24, and 28 days after inoculation. P = plasmid as positive control. H2O = nuclease-free water as negative control. M = DNA molecular marker.
The PRSV resistance of line 474 was also measured quantitatively by qPCR (Fig. 6). Virus accumulation within plants inoculated with individual isolates F61d, F10d, and F21d, representing PRSV subgroups I, II and III, respectively, as well as with a mixture of all three isolates, was investigated 0, 12, and 24 days after inoculation. Line 474 suppressed replication of all three PRSV isolates, whether applied individually or as a mixture,, while the non-transgenic line 1280 accumulated the PRSV isolates dramatically. Virulence of the three PRSV isolates, in terms of virus accumulation within the host, was not significantly different within either line 474 or line 1280.

Quantitative real-time PCR was used to investigate the PRSV accumulation in transgenic and non-transgenic papaya leaves. Individual isolates F61d, F10d, and F21d represented PRSV Groups I, and II and III, respectively. PRSV MIX indicated the mixture of same amount of PRSV I, and II and III. The PRSV accumulation was measured at 0, 12 and 24 days after the inoculation.
Northern blotting to detect accumulation of siRNA in transgenic papaya
Evidence for PRSV resistance based on transgene silencing was strengthened by detection via Northern blotting of siRNAs in transgenic papaya seedlings (Fig. 7). Two siRNAs (<50 bp) were found in uninoculated transgenic papaya leaves. Our results also showed there were no corresponding siRNA products in non-trangenic line 1280 or traditional papaya cultivars ‘Zhongbai’ and ‘Suizhonghong 48’.

Northern blot analysis to investigate the siRNA accumulation on virus free papaya leaves (A) and virus challenged papaya leaves (B). The full-length gels are presented in Supplementary Figure 5 and Supplementary Figure 6. (A) Young tissue culture seedlings regenerated from embryogenic callus considered as virus free materials. Lane 1, 3 and 4 are non-transgenic line 1280. Lane 2, 5 and 6 are transgenic line 474. Lane 7 and 8 are traditional cultivars ‘ZhongBai’ and ‘Suizhonghong 48’ germinated from seeds as references. (B) PRSV challenged (24 days) papaya leaves were also estimated the siRNA accumulation. Lane 1 and 2 are transgenic lane 474 challenged with PRSV I isolate F61d. Lane 3 and 4 are transgenic lane 474 challenged with PRSV II isolate F10d. Lane 5 and 6 are transgenic lane 474 challenged with PRSV III isolate F21d. Lane 7 and 8 are pooled RNA mixture of non-transgenic line 1280 challenged with PRSV I, II and III respectively. Arrow indicated the probe self-hybridization dot (50 bp).
Discussion
Breakdown of transgenic PRSV resistance, which depends on sequence homology between the transgene and attacking virus strain, is a major concern facing papaya cultivation, since genetically distinct strains of PRSV have been identified throughout the world18. In Hawaii, due to geographical isolation and the relative homology of PRSV strain existing there, the transgenic resistance to PRSV introduced in papaya cultivars Rainbow and SunUp in 1998 is still effective against local strains8. However, in areas like Hainan, China, where more diverse PRSV strains exist, a narrowly based transgenic resistance is expected to drive the emergence of more virulent virus strains and eventually succumb to the adapting virus population16. Consequently, papaya cultivars with resistance to a broad spectrum of PRSV strains are urgently needed by the papaya industry in Hainan.
Other researchers have suggested introducing the HC-Pro gene into transgenic lines to overcome the potential emergence of more virulent virus strains selected by transgenic crops currently using a CP target gene11. Our approach to achieving broad transgenic resistance to PRSV involved selecting a conserved region of the CP gene as the RNAi target sequence. Analysis of 53 PRSV isolates from Hainan revealed a 544-bp region of the CP gene that shared 97 to 100% identity among all isolates.
Particle bombardment has been commonly used as a gene transformation method19,20,21. Some researchers report that particle bombardment tends to produced multiple or damaged insertions22. However, the flexibility of the particle gun method allows control of the frequency of insertions and ability to produce either low- or high-copy events by altering the quantity of cassette DNA used in the bombardment22.
The use of PTGS to produce PRSV-resistant papaya is a proven approach to management of PRSV. We used an RNAi strategy to target a conserved region of the PRSV CP gene during virus replication to develop resistance to a broad spectrum of PRSV isolates from Hainan. Our results showed that transgenic line 474 produced two siRNAs with molecular weights of less than 50 bp. In short term (4-week) greenhouse experiments, transgenic line 474 showed resistance to PRSV strains that caused disease symptoms in non-transgenic controls, but evaluation of resistance durability must await longer term field trials using inoculated and uninoculated plants grown from seedling generations (Supplementary Figure 8).
Methods
Plant materials
Traditional papaya cultivars ‘Sunrise’, ‘Zhongbai’ and ‘Suizhonghong 48’ were collected in the lab. Genomic DNA from papaya cultivar ‘SunUp’ was provided by Hawaii Agriculture Research Center, Waipahu, HI. USA.
Embryogenic callus tissue from seeds of the papaya cultivar ‘Sunrise’ was cultured on the induction medium23. Papaya transformation and regeneration were carried out by particle bombardment as described previously24.
PRSV isolates and inoculation bioassay
The PRSV isolates from Hainan province are divided into subgroup I, II and III16. Isolates F61d, F10d, and F21d from each subgroup, respectively, were maintained −80 °C. Virus inoculum was prepared by inoculating local papaya plants ‘Suizhonghong 48’ prior to each bioassay. Papaya leaf samples with the well-developed symptoms of PRSV were homogenized with phosphate buffer solution (PBS) 1:10 (w/v), followed by centrifugation at 6000 × g, 4 °C. The supernatant was used as inocula within 48 hours or stored at −80 °C.
Transgenic line and non-transgenic papaya plants with four to six leaves were used to evaluate plant resistance. The third or fourth leaf from the top was gently rubbed with quartz sand using a blade tip. A 10-μl aliquot of inoculum was applied to each rubbed spot. Inoculum stayed on the leaf surface 5 min prior to washing with distilled water. PBS buffer was used as a control inoculum. All inoculated plants were kept in a growth chamber with 16 h light at 28 °C and 8 h dark at 25 °C. The rate of infection from two independent tests, each with 12 plants per line, was statistically analyzed using Duncan’s multiple range tests. Individual isolates as well as a mixture of isolates F61d, F10d, and F21d were used to inoculate transgenic line and non-transgenic papaya line.
Vector construction and plant transformation
The genetic variability among PRSV isolates in Hainan was investigated in a previous study16. In this study, in order to use hairpin construct to generate a higher efficiency of RNA silencing, we selected the conserved region of the CP genes of the different Hainan isolates as the RNAi target sequence (Supplementary Figure 1). Sequence analysis of the 544-bp region of the Hainan PRSV isolates shared 97 to 100% identity.
The conserved fragment of PRSV CP gene was used to design RNAi hairpin structures. The constructed CP hairpin structure was inserted into pCAMBIA2300-35S-OCS to construct the plant expression RNAi vector (Fig. 1).
The total RNA from papaya leaves infected with isolate F61d (PRSV I) was extracted with RNeasy Plant Mini Kit (QIAGEN). The first strand of cDNA was synthesized by using the ImProm-II Reverse Transcription System (Promega). The targeted CP fragment with restriction sites (Table 1) was amplified by high-fidelity DNA polymerase (NEB). Two “directional” CP fragments (CP sense strand was digested with Xho I and Bgl II, and CP reversed strand was digested with Sal I and BamH I) were ligated to pUCCRNAi plasmid25 to form the hairpin structure with a 201-bp intron. The hairpin structure was digested with Sal I and Pst I and inserted into pCAMBIA2300-35S-OCS to form the plant expression vector (Fig. 1 and Supplementary Figure 2)26.
Plasmid DNA was applied to 1.6 μm gold particles (Bio-Rad, Hercules, Calif.) that were inserted into embryogenic callus using a PDS 1000 Helium (Bio-Rad) particle bombardment system as previously described24. Each target plate, containing approximately 1 g fresh weight of embryogenic callus, was bombarded three times. The bombarded callus was placed on half-strength MS medium, pH 5.8, containing 10 mg/L 2, 4-dichlorophenoxyacetic acid, 3% sucrose, and 2.5% Phytagel for 10 days to recover, and then treated with 100 mg/L Kanamycin.
PCR validation the transgenic lines
PCR was used to detect transformation events in plants regenerated from the callus lines. DNA was extracted from 100 mg of papaya leaves using the E.Z.N.A. High Performance (HP) Plant DNA Kit (Omega) according to the manufacturer’s instructions. We used target-gene-specific primers, CP1/CP2, and construct-specific primers, 35S-F/CP2, to detect inserted sequences, and endogenous papain gene primers, papainF/papainR, as a check for proper function of PCR assays (Table 1). PCR was performed on a thermocycler (Biometra) as follows: one cycle of initial denaturation at 94 °C for 3 min; 35 cycles of denaturation at 94 °C for 30 s, annealing at 58 °C for 30 s and extension at 72 °C for 30 s (60 s for the 35S-F/CP2 primers); and one cycle of final extension at 72 °C for 10 min.
Two primers, 474-61 F/474-61 R, were designed to amplify a 192-bp sequence of papaya DNA centered on the transgene insertion site of line 474. A nested PCR assay containing both the insert site-specific primers, 474-61 F/474-61 R, and transgene-specific primers, CP1/CP2, was designed to confirm transgenic lines. The amplification cycling program consisted of one cycle of initial denaturation at 94 °C for 3 min; 35 cycles of denaturation at 94 °C for 30 s, annealing at 58 °C for 30 s and extension at 72 °C for 2 min; and one cycle of final extension at 72 °C for 10 min.
The QX100TM Droplet Digital PCR system (ddPCR, Bio-Rad, USA) was used with target-gene primers CP1/CP2 referenced to the endogenous papain gene to estimate the target-gene copy number in transgenic lines 474 and SunUp, following the manufacturer’s instructions. The ddPCR amplification cycling program began with an initial denaturation at 95 °C for 5 min, followed by 45 cycles of denaturation at 95 °C for 20 s, annealing and elongation at 58 °C for 30 s; and the final cycle of incubation at 98 °C for 10 min. QuantaSoft (V1.3.2.0) software was used to analyze the experimental data and quantify the PCR products. Three biological replicates per transgenic genotype x primer-set treatment, and three technical replicates per biological sample, were assayed.
Southern blot analysis to confirm transgenic events
Papaya DNA was extracted with the E.Z.N.A. High Performance Plant DNA Maxi Kit (Omega) and used for Southern blotting. CP-gene probes were prepared by amplification with specific primers CP1 and CP2 (Table 1) and labeled with Digoxigenin-11-dUTP using DIG-High-Prime Kit (Roche). About 60 μg of genomic DNA was digested with Hind III and BamH I, separately. Electrophoresis of the genomic DNA was performed in 1.0% agarose gel. The DNA was transferred to nylon membranes (GE Healthcare) using the capillary siphon blot method27, and then fixed by UV-crosslinking (Scientz Biotechnology, LTD) at 243 nm for 45 min. Hybridization was performed overnight at 42 °C in a hybridization incubator (Model 2000 Robbins Scientific). Immunological detection was made with anti-Digoxigenin-AP following the product manual, and recorded with an EC3 imaging system (UVP, LLC).
Northern blot analysis to investigate siRNA accumulation
Tissue cultured transgenic and non-transgenic papaya plants, virus free or 24 days after inoculation with PRSV, provided leaf RNA extracts via RNeasy Plant Mini Kit (QIAGEN) for investigation of siRNA accumulation. The integrity of RNA was checked with formaldehyde denaturing agarose gel electrophoresis27. The conserved 544-bp sequence of the PRSV CP gene was fragmented into 11 probes averaging 50 bp in length, of which probe CP544-10 (from 451 bp to 500 bp) and probe CP544-11 (from 501 bp to 544 bp) were later confirmed to produce efficient hybridization signals (Table 1). The Northern blot probes were labeled with Digoxigenin-11-dUTP using DIG-High-Prime Kit. Both 15% formaldehyde denaturing agarose gels (lab made) and 15% Mini-PROTEAN TBE-Urea Gels (Bio-Rad, USA) were used to separate the small RNAs by loading approximately 60-80 μg of RNA per sample, and transferring the separated bands to nylon membranes (GE Healthcare) using the capillary siphon blot method27. Hybridization was performed overnight at 42 °C in a hybridization incubator (Model 2000, Robbins Scientific). Immunological detection was the same as for the Southern blot, except that the containers, reagents and buffers were pre-treated with 0.1% DEPC (v:v) water to remove contaminating RNAase.
High-efficiency thermal asymmetric interlaced PCR (hiTAIL-PCR)
The candidate transgenic lines were screened for insertion sites using the hiTAIL-PCR method28. The primers used are listed in Table 1. PCR products were cut from the agarose gel and purified with the E.Z.N.A. Gel Extraction Kit (Omega). The purified amplicons were cloned in a pMD18 vector (TaKaRa) following the manufacturer’s protocol, and then transformed into Escherichia coli DH5α competent cells. Three positive colonies from each transformation were sequenced with an ABI 3130xl Genetic Analyzer (Hitachi), and a result of identical sequences from all three colonies was considered confirmation of the actual sequence.
RT-PCR and qPCR to detect PRSV-resistant lines
PRSV accumulation in leaves of transgenic and non-transgenic papaya plants was investigated after inoculation with individual PRSV isolates (F61d, F10d, or F21d), as well as a mixture of all three isolates. Each papaya genotype x PRSV isolate treatment had 5 biological replicates. The tissue cultured papaya plants were cultivated in the greenhouse and observed every four days for symptom development.
PRSV accumulation in papaya leaves after inoculation with the mixture of PRSV isolates was monitored in a time series of 0, 4, 8, 12, 16, 20, 24 and 28 days after inoculation (DAIs). For this assay, we used the PRSV HC-Pro gene as an indicator of the virus accumulation. Plant RNA was extracted with an RNeasy Plant Mini Kit (QIAGEN). First-strand cDNA was synthesized using the ImProm-II Reverse Transcription System (Promega) by starting with total RNA and oligo (dT)15. HC-Pro gene-specific primers were used for second-strand amplification and were able to amplify PRSV I, II and III (Table 1). The polymerase chain reaction (PCR) mixture contained 1 µl of cDNA, 0.5 µl of each primer (10 µM), 2 µl of 10 × PCR buffer, 1 µl of dNTP, 0.25 U of Taq DNA polymerase (Takara), and nuclease-free water to a final volume of 20 µl. The amplification consisted of 35 cycles at 94 °C (30 s), 50 °C (1 min), and 72 °C (1 min); the initial cycle incorporated a melting step of 94 °C for 3 min and the final cycle a synthesis step of 72 °C for 10 min. PCR products were electrophoresed on 1.5% agarose gel, and visualized after ethidium bromide staining.
Quantitative real-time PCR investigated the PRSV accumulation in papaya leaves at 0, 12 and 24 days after the inoculation with individual isolates (F61d, F10d, and F21d) and PRSV mix. A Mx3005 P Thermocycler (Strata Gene) was used in the amplification. The accumulation of PRSV was detected by SYBR Premix Ex Taq (TaKaRa) with signal correction using ROX reference dye.
Change history
22 November 2017
A correction to this article has been published and is linked from the HTML version of this paper. The error has been fixed in the paper.
References
Gonsalves, D., Tripathi, S., Carr, J. B. & Suzuki, J. Y. Papaya Ringspot virus. The Plant Health Instructor, https://doi.org/10.1094/PHI-I-2010-1004-01 (2010).
Olarte Castillo, X. A. et al. Phylogeography and molecular epidemiology of Papaya ringspot virus. Virus research 159, 132–140, https://doi.org/10.1016/j.virusres.2011.04.011 (2011).
Gonsalves, D. Control of papaya ringspot virus in papaya: A case study. Annual Review of Phytopathology 36, 415–437 (1998).
Purcifull, D. E. & Hiebert, E. Serological distinction of Watermelon mosaic virus isolates. Phytopathology 69, 112–116, https://doi.org/10.1094/phyto-69-112 (1979).
Reddy, P. P. In Sustainable Crop Protection under Protected Cultivation 161–176 (Springer Singapore, 2016).
Ming, R. et al. The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452, 991–996, https://doi.org/10.1038/nature06856 (2008).
Porter, B. W. et al. Genome-wide analysis of Carica papaya reveals a small NBS resistance gene family. Molecular Genetics and Genomics 281, 609–626, https://doi.org/10.1007/s00438-009-0434-x (2009).
Gonsalves, D. Control of papaya ringspot virus in papaya: a case study. Annual review of phytopathology 36, 415–437, https://doi.org/10.1146/annurev.phyto.36.1.415 (1998).
You, B. J., Chiang, C. H., Chen, L. F., Su, W. C. & Yeh, S. D. Engineered Mild Strains of Papaya ringspot virus for Broader Cross Protection in Cucurbits. Phytopathology 95, 533–540, https://doi.org/10.1094/PHYTO-95-0533 (2005).
Ferreira, S. A. et al. Virus Coat Protein Transgenic Papaya Provides Practical Control of Papaya ringspot virus in Hawaii. Plant Disease, 101–105 (2002).
Kung, Y. J. et al. Nucleotide sequence-homology-independent breakdown of transgenic resistance by more virulent virus strains and a potential solution. Sci Rep 5, 9804, https://doi.org/10.1038/srep09804 (2015).
Tennant, P. F. et al. Differential Protection Against Papaya Ringspot Virus Isolates in Coat Protein Gene Transgenic Papaya and Classically Cross-Protected Papaya. Phytopathology 84, 1359–1366 (1994).
Kung, Y. J. et al. Generation of transgenic papaya with double resistance to Papaya ringspot virus and Papaya leaf-distortion mosaic virus. Phytopathology 99, 1312–1320, https://doi.org/10.1094/PHYTO-99-11-1312 (2009).
Bau, H. J., Cheng, Y. H., Yu, T. A., Yang, J. S. & Yeh, S. D. Broad-Spectrum Resistance to Different Geographic Strains of Papaya ringspot virus in Coat Protein Gene Transgenic Papaya. Phytopathology 93, 112–120, https://doi.org/10.1094/PHYTO.2003.93.1.112 (2003).
Davis, M. J. & Ying, Z. Development of Papaya Breeding Lines with Transgenic Resistance to Papaya ringspot virus. Plant Disease 88, 352–358 (2004).
Zhao, H. et al. Geographical and Genetic Divergence Among Papaya ringspot virus Populations Within Hainan Province, China. Phytopathology 106, 937–944, https://doi.org/10.1094/PHYTO-05-15-0111-R (2016).
Giladi, E., Raz, E., Karmeli, F., Okon, E. & Rachmilewitz, D. Transforming growth factor-beta gene therapy ameliorates experimental colitis in rats. European journal of gastroenterology & hepatology 7, 341–347 (1995).
Gonsalves, D. Transgenic papaya: development, release, impact and challenges. Advances in virus research 67, 317–354, https://doi.org/10.1016/S0065-3527(06)67009-7 (2006).
Sparks, C. A. & Jones, H. D. Genetic transformation of wheat via particle bombardment. Methods in molecular biology 1099, 201–218, https://doi.org/10.1007/978-1-62703-715-0_17 (2014).
Liu, G., Campbell, B. C. & Godwin, I. D. Sorghum genetic transformation by particle bombardment. Methods in molecular biology 1099, 219–234, https://doi.org/10.1007/978-1-62703-715-0_18 (2014).
Liu, C. W., Lin, C. C., Chen, J. J. & Tseng, M. J. Stable chloroplast transformation in cabbage (Brassica oleracea L. var. capitata L.) by particle bombardment. Plant cell reports 26, 1733–1744, https://doi.org/10.1007/s00299-007-0374-z (2007).
Lowe, B. A. et al. Enhanced single copy integration events in corn via particle bombardment using low quantities of DNA. Transgenic research 18, 831–840, https://doi.org/10.1007/s11248-009-9265-0 (2009).
Kung, Y. J. et al. Generation of hermaphrodite transgenic papaya lines with virus resistance via transformation of somatic embryos derived from adventitious roots of in vitro shoots. Transgenic research 19, 621–635, https://doi.org/10.1007/s11248-009-9344-2 (2010).
Fitch, M. M. & Manshardt, R. M. Somatic embryogenesis and plant regeneration from immature zygotic embryos of papaya (Carica papaya L.). Plant cell reports 9, 320–324, https://doi.org/10.1007/BF00232860 (1990).
Gan, D. et al. Bacterially expressed dsRNA protects maize against SCMV infection. Plant Cell Reports 29, 1261–1268, https://doi.org/10.1007/s00299-010-0911-z (2010).
Zhao, H. et al. Construction of RNAi Plant Expression Vectors with Broad Spectrum Resistance to Hainan Papaya Ringspot Virus. Chinese Journal of Tropical Crops 36, 2210–2215 (2015).
Sambrook, J. & Russell, D. W. Molecular Cloning: A laboratory manual. 3rd ed. edn, (Cold Spring Harbor Laboratory Press, 2001).
Liu, Y. G. & Chen, Y. High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences. BioTechniques 43, 649–650, 652, 654 passim (2007).
Acknowledgements
We thank Dr. Dennis Gonsalves from the USDA-ARS Pacific Basin Agricultural Research Center, Dr. Richard M. Manshardt and Dr. John Hu from the University of Hawaii, and Dr. Shyi-Dong Yeh from Chung-Hsing University, Taiwan, for sharing their experiences with transgenic papaya. Our work was supported by the International Advanced Agricultural Science and Technology Projects of China, Ministry of Agriculture (2014-Z17). This work was also supported by the Central Public Fundamental Scientific Research, Ministry of Agriculture (1630052016019 and 1630052016001), by the Hainan Provincial Key Research and Development Plan (ZDYF2016027), and by the Chinese Academy of Tropical Agricultural Sciences Innovation Team (17CXTD-24).
Author information
Authors and Affiliations
Contributions
Ruizong Jia, Hui Zhao, Yun J. Zhu, Ming Peng and Anping Guo conceived and designed the experiments; Jing Huang and Ruizong Jia performed molecular characterization of the transgenic event; Ruizong Jia, Hui Zhao and Hua Kong completed the PRSV survey; Yuliang Zhang and Qixing Huang constructed the vector; Ruizong Jia and Qing Wei prepared the papaya callus and were responsible for tissue culture; Hua Kong, Hui Zhao, Ruizong Jia and Yun J. Zhu performed the genetic transformation. Ruizong Jia, Hui Zhao, Yunling Guo, Jingyuan Guo, and Jiao Zuo carried out the selection. Ruizong Jia and Jing Huang analyzed the data; Ruizong Jia and Jing Huang wrote the paper.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Change History: A correction to this article has been published and is linked from the HTML version of this paper. The error has been fixed in the paper.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A correction to this article is available online at https://doi.org/10.1038/s41598-017-16198-4.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jia, R., Zhao, H., Huang, J. et al. Use of RNAi technology to develop a PRSV-resistant transgenic papaya. Sci Rep 7, 12636 (2017). https://doi.org/10.1038/s41598-017-13049-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-13049-0
This article is cited by
-
Evolution of plant mutagenesis tools: a shifting paradigm from random to targeted genome editing
Plant Biotechnology Reports (2019)
-
Transgene-mediated resistance to Papaya ringspot virus: challenges and solutions
Phytoparasitica (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
