
Abstract
Research on toxin-antitoxin loci (TA loci) is gaining impetus due to their ubiquitous presence in bacterial genomes and their observed roles in stress survival, persistence and drug tolerance. The present study investigates the expression profile of all the seventy-nine TA loci found in Mycobacterium tuberculosis. The bacterium was subjected to multiple stress conditions to identify key players of cellular stress response and elucidate a TA-coexpression network. This study provides direct experimental evidence for transcriptional activation of each of the seventy-nine TA loci following mycobacterial exposure to growth-limiting environments clearly establishing TA loci as stress-responsive modules in M. tuberculosis. TA locus activation was found to be stress-specific with multiple loci activated in a duration-based response to a particular stress. Conditions resulting in arrest of cellular translation led to greater up-regulation of TA genes suggesting that TA loci have a primary role in arresting translation in the cell. Our study identifed higBA2 and vapBC46 as key loci that were activated in all the conditions tested. Besides, relBE1, higBA3, vapBC35, vapBC22 and higBA1 were also upregulated in multpile stresses. Certain TA modules exhibited co-activation across multiple conditions suggestive of a common regulatory mechanism.
Similar content being viewed by others

Introduction
Mycobacterium tuberculosis, the causative pathogen of the deadly disease tuberculosis, has evolved mechanisms to persist in the human host for long times in a non-replicating and drug-tolerant state, and reactivate at a later time to cause disease. The underlying mechanisms and cellular factors that govern the transition of the bacterium from an active state to latent state are poorly understood1,2.
Recent studies in Escherichia coli and other bacteria point out a prominent role for toxin-antitoxin (TA) systems in stress survival and bacterial persistence3,4,5,6,7. TA loci are typically two-component systems that consist of a stable toxin and a relatively unstable antidote of the toxin, the antitoxin, both being encoded as an operon8. In most cases, the operon is auto-regulated at the level of transcription by the antitoxin alone and in some cases by the toxin-antitoxin complex9,10,11. Degradation of the labile antitoxin serves as a mechanism for transcriptional induction of TA locus genes as well as for the availability of free toxin protein to act on its cellular target. The most common targets of TA-encoded toxins are translation components; some others have been shown to target proteins belonging to the replication machinery12,13,14,15,16,17,18,19. TA-encoded toxins act to rapidly slow down cellular processes, giving bacteria an opportunity to alter their metabolic program and enter into a dormant or non-replicating state and exhibit a persistent or drug-tolerant phenotype4,6,7,20,21,22. Cells unable to adapt to this metabolic slow down initiate a programmed cascade of events leading to cell death23,24,25.
Thirty-eight TA loci were initially reported in M. tuberculosis26, however, more are being identified27. Presently, M. tuberculosis is reported to have 79 TA loci spanning the entire genome28. Several reports have been published on the characterization of these loci, identification of the target sites for toxins, structural characterization of some antitoxins, evaluation of their killing effect in E. coli and expression under stress27,29,30,31,32,33,34,35,36,37,38,39. Stochastic induction of specific TA systems has been proposed to play a role in persistence of M. tuberculosis in the host40,41,42.
Considering the fact that M. tuberculosis harbors an unusually high number of TA loci for any bacterial genome and more importantly, significantly higher than other closely related mycobacteria like M. avium, M. bovis and M. smegmatis, it will be useful to analyze the expression profiles of TA genes under different stress conditions thought to mimic the host environment during mycobacterial infection to elucidate their contribution towards the survival ability of this deadly pathogen. While ectopic expression of certain toxins has been shown to inhibit growth of M. tuberculosis in vitro and certain TA loci get induced under specific stress31,32,35,43, a comprehensive profile of genome-wide TA loci of M. tuberculosis is lacking. A system-wide screening will help identify specific TA loci differentially expressed under different growth conditions and also shed light on co-regulated pathways/genes that could provide clues to mechanisms for TA activation and regulation.
We have developed a whole-genome microarray platform for M. tuberculosis that harbors specific probes for all the 79 TA loci44. Prior published microarrays for M. tuberculosis did not contain probes for all these 79 TA loci. In the present study, we elucidate TA expression patterns in response to chemical and environmental stress and identify key TA loci that could have potential roles in the pathogenesis of M. tuberculosis. This is the first study covering various stress conditions across multiple time points on a single platform with key emphasis on genome-wide TA loci of M. tuberculosis.
Results
Expression profiling of TA loci in M. tuberculosis
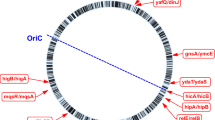
TA loci identified in various bacterial genomes are grouped into families based on sequence homology and biochemical properties26. M. tuberculosis H37Rv genome harbours 79 TA loci spanning the entire genome (Fig. 1) of which 68 loci have been assigned to the existing TA families. There are three higBA loci, ten mazEF loci, two parDE loci, three relBE loci, and fifty vapBC loci. The remaining 11 loci (ucAT) are uncharacterised and yet to be assigned to existing/novel TA families.

Toxin Antitoxin loci encoded by Mycobacterium tuberculosis H37Rv The 79 TA loci present in M. tuberculosis H37Rv genome tabulated with the locus name, gene name for toxin and antitoxin, gene identifier (Rv number), genomic location as Start base position and End base position, orientation in the genome and the type of TA locus. The uncharacterized TA loci are given the nomenclature as ucAT followed by a number for the series. The number in the blue bar indicates the total number of experimental conditions wherein differential expression was observed for the toxin gene and the number in the pink bar indicates the same for the antitoxin gene.
Using a recently described M. tuberculosis microarray covering all these 79 TA loci44, we obtained the expression profile of M. tuberculosis H37Rv after subjecting it to various stresses including chemical (drug) stress and environment stress (nutrient starvation and low pH). The four frontline drugs that comprise TB chemotherapy namely Rifampicin (RIF), Isoniazid (INH), Ethambutol (ETH) and Streptomycin (STR) were used in this study to understand changes in expression pattern of TA loci in response to drug administration to the bacteria. Low pH shift was used to mimic environment of mycobacteria in the phagosomal compartment of the host macrophage and starvation (STRV) was used to mimic the nutrient limitation encountered by the bacterium in the host. M. tuberculosis was subjected to these stresses and samples at multiple time points were used for expression studies. Quantitative RT-PCR was performed to check expression of selected marker genes and validate the cultures for stress exposure. These marker genes were selected based on published literature45,46. The fold change obtained for various genes correlated with published data for different conditions (data not shown). Some of these conditions have been used previously to define the transcriptome of M. tuberculosis by Boshoff et al.45. They have analysed the expression profile of M. tuberculosis H37Rv under different growth conditions in culture. However, their microarray design does not contain probes for all the 79 TA loci nor does their analysis focus on the expression of TA loci under different conditions. Our study aims to address this gap in existing knowledge by providing data and its analysis for all the 79 TA loci.
After transcriptional profiling of all the samples representing eighteen stress conditions, the data obtained was analysed for differentially expressed genes. As shown in Fig. 1, each of the 79 TA locus exhibits differential expression in M. tuberculosis in response to one or more stress conditions studied here, signifying the need of the cell to alter the expression of TA genes in response to stress. Further, most genes have altered expression across multiple stress conditions. However, no one gene is expressed under all the conditions tested. Even members from within the same TA family are differentially expressed under different set of conditions. This clearly suggests a stress-specific expression of TA genes. higBA1, higBA2, higBA3, mazEF1, mazEF6, mazEF7, parD1, parD2, relBE3, vapBC5, vapBC6, vapBC14 and vapBC21 loci had differential expression of their component genes in at least 50% of the stress conditions studied. Some of the uncharacterised TA loci, namely ucAT2, ucAT9 and ucAT11 were also expressed under multiple stress conditions. It should be noted that these 158 toxin and antitoxin genes out of an approximately 4000 total genes in the mycobacterial genome comprise 4% of the coding capacity of the bacterium and belong to a single stimulus-responsive system, the TA system. This accumulation and conservation of TA loci in large numbers uniformly across the genome and their induction in response to varied environment changes is clearly indicative of their role in increasing the pathogenic fitness of mycobacteria and contributing to its ability to survive in different niches in the host. This also emphasises the need for maintenance of TA loci in a bacterial genome and their ubiquitous presence across the bacterial kingdom.
Stress-induced differential expression of TA loci in M. tuberculosis
Variation in copy number of expressed transcripts leads to differences in sensitivity of detection in a microarray platform. In order to comprehensively delineate the differentially expressed TA genes in a sensitive manner, we analyzed the data with variable Fold Change (FC) cut-off at 5% confidence level. The total number of differentially expressed TA genes increased with the duration of the stress for each of the conditions (Fig. 2), suggestive of their essential and growing requirement in stress response. The greatest up-regulation of TA genes was observed in response to STR and nutrient starvation followed by RIF and INH treatments. Shift to low pH resulted in more downregulation of TA genes than upregulation. The number of differentially expressed TA genes reduced when the cutoff was raised from 1.5 to 2.0, however, greater than 50% TA genes were still differentially expressed (Supplementary Table S1). The expression pattern at 1.5 FC and 2.0 FC followed a similar trend (Fig. 2), indicating sensitivity of detection of the microarray platform. Moreover, though the number of genes was higher at lower Fold Change as expected, the TA genes identified were truly differentially expressed. The microarray data were validated using qRT-PCR for some of the TA loci. A strong positive correlation (Pearson’s correlation coefficient, R = 0.897) was observed between the microarray and qRT-PCR data (Supplementary Fig. S1).

Differentially expressed TA Loci across experimental conditions. Line Graph depicts the number of TA genes that are upregulated and downregulated in each experimental condition at 1.5 and 2.0 fold change. Total number of up and down regulated toxin and antitoxin genes for each condition were plotted using Microsoft excel. The Y-axis indicates total number of toxin and antitoxin genes differentially regulated and X-axis indicates the experimental conditions and time points of the study. Ethambutol (ETH), Isoniazid (INH), Rifampicin (RIF), Streptomycin (STR), Starvation (STRV). 4 H, 6 H, 24 H, 72 H denotes the duration of treatment as 4 hours, 6 hours, 24 hours and 72 hours. 1WK denotes treatment for 1 week.
Differentially expressed TA genes unique to a given condition and those expressed across multiple conditions were identified using the matrix shown in Fig. 3. An interactive matrix showing the differentially expressed gene list for each condition is given in Supplementary Fig. S2. At 1.5 FC, 8 TA genes (vapC2, vapC32, mazE4, mazE5, mazE7, vapB38, vapC38 and vapB19) were uniquely upregulated in response to exposure to Streptomycin for 72 hours (Fig. 3A). These were not common with any other condition tested. Similarly, vapB18 was specifically upregulated in response to exposure to Ethambutol for 72 hours and UCT7 in response to Rifampicin for 72 hours. Two TA genes, vapB44 and ucA5, were specifically upregulated in response to nutrient starvation for 24 hours time period. 50% TA genes were common between Streptomycin and starvation stress, indicating that both the conditions induced similar TA responses in the cell. Several TA genes were found to be common under different stress conditions. Significantly higher numbers of TA genes were upregulated than downregulated under any given condition, supporting the role of TA loci as stress managers in the cell. At 2.0 FC (Fig. 3B and D), more TA genes unique to a given condition were identified. Here also, up-regulation of TA genes in response to streptomycin and nutrient starvation was maximal.

Differential gene expression matrix showing the number of up and down regulated TA genes across the experimental conditions. TA genes up and down regulated at varying sensitivity of detection were subjected to GeneMatrix software which resulted in distribution of up and down regulated genes across 18 conditions profiled. The upregulated genes at 1.5 FC (A) and 2.0 FC (B) are shown in increasing color of red while the downregulated genes at 1.5 FC (C) and 2.0 FC (D) are shown in green.
Analysis of the matrix suggests that TA gene transcription is under tight regulation. A marginal change in sensitivity of copy-number of detection by increase in FC from 1.5 to 2.0 leads to a significant drop in the number of differentially expressed TA genes, indicating a likely stringent regulatory control on gene expression. At the same time, a reduction in FC does not substantially increase the number of condition-specific genes, validating that genes identified specific to a given condition are truly specific.
Unsupervised hierarchical cluster analysis of differentially expressed TA loci under different conditions was carried out (Fig. 4). Here again, the TA response clustered nutritional starvation with prolonged exposure to Streptomycin, indicating that the two stress conditions activate similar responses and pathways. The cell wall inhibitors, INH and ETH clustered together while the transcription inhibitor RIF gave a different response. Further, the 72 hours response clustered separately from the shorter duration response signifying the need of the cell to alter the expression of TA genes in response to the duration of stress. The drug–induced response clearly clustered separate from the pH stress-induced response in line with the earlier observation wherein pH was found to significantly down-regulate TA gene expression in contrast to other conditions. The cluster analysis highlights the diversity and division of labour in a biological organisation wherein even a small component like TA system gives a differential, stimulus- specific response.

Unsupervised hierarchical clustering analysis of Differentially expressed TA Loci TA genes 1.5 fold up and down regulated along with their fold change were subjected to unsupervised hierarchical clustering. Up regulated genes are in red color gradient and down regulated genes are in green colour gradient. Fold change of 2 was given as a cutoff to render the cluster image to identify patterns of up and down regulated genes.
Impact of frontline TB drugs on transcription of TA loci
The four anti-TB drugs used in this study target different cell components and pathways. We performed an in-depth study of the M. tuberculosis transcriptome for cells treated with these drugs for three time points, to capture the primary effect of the drug as well as extended treatment response.
Treatment with Streptomycin had the most profound effect on TA expression. Streptomycin interferes with several steps of protein synthesis, its most conspicuous effect being the stimulation of translational errors and a slowing down of translocation resulting in the production of faulty proteins. A large number of TA loci were differentially regulated in response to this drug. Most loci were upregulated with increased duration of drug exposure (Fig. 4 and Supplementary Table S1). The toxin as well as antitoxin genes were upregulated for all the three higBA loci with progressively increasing levels. The toxin partner showed higher transcript levels compared to the cognate antitoxin. higB1 toxin showed a 25-fold increase, higB2 showed a 23-fold increase and higB3 showed a 10-fold increase in expression levels following exposure of the cell to STR for 72 hours. The toxin component of the uncharacterized TA locus ucAT9 showed a 21-fold increase in transcript levels. Both genes comprising relBE3, vapBC25 and vapBC31, vapBC36, vapBC41, vapBC45 and vapBC46 loci were also upregulated 2–10 fold at 24 hrs post-drug exposure. At 72 hrs of exposure to STR, 25 loci were upregulated. For some of these loci including mazEF5, mazEF8, vapBC18, vapBC20, vapBC24 and vapBC33, the toxin was upregulated while the antitoxin partner gene showed less or no upregulation.
Rifampicin, another frontline TB drug acts on the mycobacterial RNA polymerase leading to inhibition of transcription. The expression profile of TA loci in response to Rifampicin, across three time points is shown in Fig. 4. Rifampicin exposure induced differential expression of several TA loci. A total of 26 genes were upregulated and 22 genes were downregulated. At 4 hrs post-exposure, minimal changes in expression of TA loci were observed with vapBC16, vapBC23 and vapBC49 showing a 2–4 fold upregulation. However, by 24 hrs of drug exposure, a substantial number of TA genes were differentially expressed with several of them being upregulated 2–4 fold. higBA3, mazEF1, mazEF6, mazEF9, parDE2, relBE1, relBE2, ucAT1, ucAT2, ucAT10, ucAT11, vapBC1, vapBC3, vapBC4, vapBC8, vapBC11, vapBC13, vapBC16 and vapBC39 showed up-regulation of both partners at 24 hrs. vapC11, vapC24, and vapC40 toxins were preferentially upregulated compared to their antitoxin counterparts. vapBC10, vapBC14, vapBC22, vapBC27, vapBC44, vapBC47 and vapBC50 were downregulated 4–10 fold across all time-points. In an earlier study, Singh et al.36 have also reported significant upregulation of relB1 and relB2 antitoxins following exposure to Rifampicin. Their results corroborate with our observations.
The changes in expression profiles of TA loci in response to Ethambutol and Isoniazid, both of which target the cell wall system, were analyzed. While there was no significant change in TA gene expression at 6 hours of drug treatment, differential expression was observed 24 and 72 hours post-drug exposure. vapC7, vapB29, vapC30, vapB4, vapC4, vapB43, vapB49 and vapC49 were upregulated 2 fold in response to Ethambutol exposure for 72 hours while vapBC44, vapC45 and vapC47 were down regulated. While the fold change in expression levels was only 2 fold for most loci, ucAT9 showed a 13-fold upregulation for the antitoxin gene and a 5-fold upregulation for the toxin gene, clearly emerging as a locus responsive to ETH exposure. mazE6, mazF6 and parD2 were induced in response to both Ethambutol and Isoniazid. All the three higBA loci showed a 2–4 fold upregulation in response to Isoniazid exposure. mazEF2, mazEF6, mazEF9, mazEF10, relBE2, relBE3, ucAT1, ucAT2, ucAT3, ucAT8, ucAT9, ucAT11, vapC3, vapBC4, vapBC5, vapBC7, vapBC8, vapC13, vapC20, vapC23, vapC26, vapBC28, vapBC29, vapBC30, vapB34, vapBC41, vapBC43, vapBC45 and vapBC49 were also upregulated 2–5 fold in response to Isoniazid exposure. ucA7 and vapC27 were downregulated 2–4 fold in response to Isoniazid.
Impact of environmental stress on transcription of TA loci
Nutrient starvation and acidic pH are two stressful environments encountered by mycobacteria in the host. Accordingly, the TA expression profile in response to these stress conditions was studied. We followed the Betts et al., method for starvation studies47 wherein the early exponential stage M. tuberculosis was re-suspended with PBS for starvation and monitored over 24 hrs and 1 week. Starvation led to generalized induction of TA loci, with a large number of TA loci upregulated at both 24 hrs and 1 week (Fig. 4 and Supplementary Table S1). Maximal induction was observed for higBA1, higBA2, higBA3, mazEF3, mazEF6, mazEF9, parDE1, parDE2, relBE1, relBE2, relBE3, vapBC4, vapBC9, vapBC10, vapBC12, vapBC16, vapBC17, vapBC22, vapBC23, vapBC26, vapBC28, vapBC30, vapBC32, vapBC34, vapBC35, vapBC41, vapBC45, vapBC46, vapBC49, ucAT1, ucAT2, ucAT3, ucAT8, ucAT9, ucAT10 and ucAT11. As many as twenty-eight loci were upregulated in response to starvation. Most of these TA loci encode translation-inhibiting RNases. Possibly, these toxins when activated in response to starvation, lead to inhibition of translation and selective degradation of mRNA to make nutrients available for survival under stress. ucAT9 showed a 36 fold induction after 24 hours of starvation and this increased to more than 100 fold induction after 1 week starvation. Similarly, ucAT1, higBA1, higBA2, and higBA3 showed a 10–18 fold upregulation. For higBA1, higBA2, relBE2, ucAT10, vapBC1, vapBC6, vapBC7, vapBC34, vapBC35, vapBC45 loci, the toxin was upregulated to higher levels compared to the antitoxin partner.
In contrast to starvation that led to generalized up-regulation of TA loci, low pH exposure led to down-regulation of TA genes (Fig. 4). The effect was more pronounced at pH 4.8 vis-à-vis pH 5.2. higBA2, vapBC1, vapBC4, vapBC15, ucAT10 and ucAT9 showed 2–5 fold upregulation following exposure to pH 4.8 for 72 hours. mazEF8, ucAT2, ucAT3, ucAT4, ucAT7, vapC7, vapC9, vapC20, vapC21, vapBC27, vapBC47 and vapB49, showed 2-fold down regulation at both pH 5.2 and pH 4.8.
TA regulatory network under stress
The TA transcriptome data showed that in spite of being expressed as an operon, the antitoxin and toxin levels for a given TA locus were not same across all conditions and time points (Supplementary Table S1). Based on this, we calculated the activation/deactivation scores for TA loci as described in methods to determine condition-specific TA responses. This is diagrammatically represented in Fig. 5A. If the median fold change difference between toxin transcript and antitoxin transcript level increased with duration of stress, it was designated as TA activation representing a net increase in toxin transcript vis-à-vis antitoxin transcript. The reverse was designated as TA deactivation. STR and STRV led to activation of majority of TA loci. Bi-clustering and Hierarchical clustering of the profiles based on these scores also showed a pattern of condition-based clustering (Fig. 6). The STR and STRV response was highly similar and clustered together. Both stresses led to activation of common TA loci. Nutrient starvation has been shown to cause metabolic slowdown in cells to economize on available resources, which results in arrest of translation, a phenomenon similar to Streptomycin action. The common response in the two conditions and the greater up-regulation of TA genes suggests that TA loci have a primary role in arresting translation in the cell. This is in line with the suggested mechanisms of TA loci wherein several of them are known to be mRNA interferases. The profile in response to RIF was completely in contrast to the STR and STRV response. RIF exposure led to deactivation of almost 90% TA loci. The cell wall inhibitor antibiotics, namely INH and ETH clustered separately and led to activation of some common and other different TA loci. Shift to a lower pH of 5.2 did not cause substantial changes in TA loci regulation; however, shift to pH 4.8 led to a dramatic increase in the number of activated TA loci giving a response similar to STR and STRV response. This suggests that the TA activation-deactivation response is stress-related. Different chemical and environment stress trigger varied responses in the cell and lead to activation of specific sets of TA loci, which might be helping bacteria overcome the stress.

(A) Line Graph depicting the approach used to calculate activation and deactivation scores for TA loci (B) Number of TA loci that are activated, deactivated and co-expressed in each of the experimental conditions profiled. Fold change of toxin from cognate antitoxin was subtracted in early and late time point. The subtracted scores were subjected to biclustering/K-means clustering using Pearson Uncentered algorithm in GeneSpring Gx v 12.8 to provide 3 clusters of all the 78 TA loci, with clusters representing activation, deactivation and co-expression status. Similar approach was followed for each of the experimental conditions and TA loci activated, deactivated and co-expressed in each condition were identified.

Unsupervised hierarchical clustering analysis of TA loci based on their activation and deactivation status in response to stress. All the 79 TA loci with their activation-deactivation and co-expression status defined as 2 for activation, −2 for deactivation and 0 for co-expression were subjected to unsupervised hierarchical clustering using Cluster 3.0 software by applying Pearson Uncentered algorithm with average linkage rule. The resultant cluster file was imported into Java Treeview software to visualize the cluster. Activated TA loci are in red color gradient and Deactivated TA loci are in green color gradient. A score of 2 was given as a cutoff to render the cluster image to identify patterns of activation and deactivation of TA loci across conditions profiled.
Cluster analysis further showed that certain TA loci were co-activated under different stress conditions. higBA1, mazEF2, mazEF5, parDE2, relBE2 and ucAT4 were always activated and deactivated under same conditions indicating that they had common regulatory mechanisms. Similarly, mazEF7, mazEF8 and ucAT8 were co-regulated and clustered together. ucAT9 and ucAT11 showed a completely different pattern of regulation and formed a separate cluster from the other loci. Perhaps different sets of transcription factors responded to different stimuli and regulated different groups of TA loci.
The data were further plotted to create a TA activation-deactivation network (Fig. 7). All the TA loci were mapped based on their activation and deactivation scores with the intensity of the color correlating to the scores and the diameter of the circle corresponding to the number of conditions in which activation-deactivation was observed. As shown, there is a clear grouping of loci that are activated from those that are deactivated. vapBC40, higBA2 and vapBC46 were highly activated in all the conditions except low pH 5.2. relBE1, vapBC35, vapBC22, vapBC2 were also upregulated in 4 out of 6 stresses, however, the activation score was less than that for higBA2 and vapBC46. mazEF9 was activated in 2 conditions and deactivated in 4 conditions. ucAT11 was deactivated in all the 6 conditions studied. Other loci including vapBC41, ucAT6, ucAT9 and higBA3 were downregulated in 5 out of the 6 conditions. vapBC13 was uniquely activated in response to INH and ETH treatment while parDE2 was activated under INH stress. Several other loci were activated or deactivated under one or more conditions tested. Most loci exhibited an activation or deactivation trend across all conditions. Only 9 out of the 79 TA loci showed a mixed trend with activation under some conditions and deactivation in other conditions.

Biological Network of TA loci. Condition-based list of TA loci activated and deactivated was provided to BridgeIsland software. This software identifies the nodes and edges that come together to form a network of connections that could be representative of the TA gene regulation. The nodes (TA loci) are colored based on the activation and deactivation levels with activation being colored in red gradient and deactivation being colored in green gradient and co-expression colored in yellow gradient. The nodes are sized based on their connectivity scores with larger the size indicating a TA is activated/deactivated in as many conditions profiled.
The TA activation-deactivation network clearly points out a stress-responsive activation of specific TA loci and a division of labour amongst the large number of loci. Not all TA loci are activated in response to a stress and every stress does not activate the same set of TA loci. While the cellular functions are not understood for a greater number of these loci, this data clearly indicates that the large number of TA loci maintained in mycobacterial genome does not simply comprise of pseudo genes or redundancy in gene function. Rather, these loci encode functional proteins expressed in response to specific stimulus.
Expression of cellular proteases and nucleases under stress
According to the existing model, TA loci are activated during stress by degradation of antitoxin partner by cellular proteases enabling the free toxin to exert its effect48,49,50,51. In this context, we checked the expression pattern of 26 annotated mycobacterial proteases under stress. As shown in Fig. 8A, several proteases were upregulated during STR and STRV stress. The Clp family proteases that have been shown to cause antitoxin degradation in several bacteria48,50 were found to be upregulated in our study indicating that they may play similar roles in Mycobacterium tuberculosis. The protease activation pattern aptly correlates with the TA activation data wherein, many TA loci were activated during STR and STRV. Two potential proteases, Rv1577c and Rv2651c were found to be upregulated under all stress conditions except INH stress. Six of the nine putative RNases of M. tuberculosis were also activated under one or more stress conditions (Fig. 8B). Again, maximum activation was observed under STRV and low pH stress. Further studies on these gene products can elucidate their possible roles in TA activation during stress in mycobacteria.

(A) Unsupervised hierarchical clustering analysis of protease genes based on their activation and deactivation status in response to stress. (B) Unsupervised hierarchical clustering analysis of nuclease genes based on their activation and deactivation status in response to stress Fold change of genes in late time point was subtracted from early time point to calculate the activation deactivation score. The cluster was created as described for Figs 5 and 6.
Discussion
This paper is the first report providing experimental data on the expression profiles of all the seventy-nine chromosomally encoded toxin-antitoxin loci in M. tuberculosis. The data generated herein is used to understand activation and deactivation of TA loci in a time-dependent manner in response to environmental and chemical stresses and identify key players in cellular stress.
TA loci are being identified in many genomes and in greater numbers in each genome. This definitely raises curiosity about their possible roles in bacterial survival and persistence. M. tuberculosis supersedes most other bacteria for the number of TA loci it harbors in its genome. Consequently, there has been a continuing effort investigating TA loci in M. tuberculosis. Previous work from our lab and other groups has demonstrated killing activity of mycobacterial TA loci in heterologous hosts and native system27,30,31,35,39,43,52,53,54,55,56,57. Further, it has been shown that TA locus deletion strains of mycobacteria exhibit growth defective phenotype58.
To understand the conditions under which TA loci get induced in M. tuberculosis, we performed a genome-wide study and identified some key TA loci that are activated in response to chemical and/or environmental stress in M. tuberculosis. It is important to note here that all the 79 TA loci encoding 158 proteins in this study show specific differential regulation in one or more stress conditions. No random selection of 158 genes for any genome is expected to show such behavior supporting the conclusion that TA genes are stress-responsive and are helping bacteria tide over unfavorable conditions.
Our results corroborate with previous reports on expression of TA genes in M. tuberculosis31,36,59. Individual TA loci have been studied and shown to be induced during hypoxia, nitrogen limitation, starvation and drug exposure27,31,32. The vapBC locus in M. smegmatis has been implicated as playing a role in regulation of metabolic flux60. Singh et al. have also shown increased survival of relE overexpressing cells during drug treatment36. Several TA loci were identified to be upregulated in M. tuberculosis drug-tolerant persister population61. While all these studies have provided knowledge on TA loci and their activity, a comprehensive analysis of all the loci on a single platform was lacking. A genome-wide analysis of TA loci in this study has filled this gap and enabled identification of TA loci responsive to varied stress conditions. higBA2 and vapBC46 emerge as key players getting upregulated to high levels in response to chemical as well as nutritional stress. mazEF1, vapBC31, ucAT10 also are activated under both kinds of stress though to lesser levels. higBA1 and mazEF5 get activated in response to nutritional starvation and streptomycin treatment. Several of the TA loci identified to be activated under stress, like mazEF1, vapBC31, higBA1 and mazEF5 have been shown to have killing activity across mycobacterial species and even in heterologous systems27,30,36. All the three higBA loci in M. tuberculosis have been studied here and higBA2 appears to be another important locus besides higBA1 that has been the focus of earlier studies56. Many of the hitherto uncharacterised TA loci, ucAT11, ucAT6, ucAT9 show strong deactivation in response to both chemical and nutritional stress. Although individual deletions of TA genes have not been found to impair bacterial survival in a mouse infection model36, it does not rule out a role for TA loci in M. tuberculosis infection and survival in the human host.
Inhibition of translation is a key event in stress response and development of persistence and drug-tolerant phenotype in E. coli. The HigB, RelE, MazF and VapC toxins from diverse bacterial species have been identified to be mRNA interferases that cleave cellular RNA at specific sequences15,38,43,62. Transcriptomic analyis of E. coli persister cells reveals over-expression of TA modules and their encoded mRNA interferases5,6. Antibiotic and starvation stress has also been linked with expression of mRNA interferases3. The transcriptional upregulation of TA loci as observed in our study suggests a similar mechanism of translation shut-down operating in mycobacteria to impart a survival advantage to the bacteria under antibiotic pressure and nutrition deficiency. Stress-induced simultaneous activation of multiple loci would enable a rapid shutdown of cellular machinery and metabolic slowdown leading to a dormant phenotype.
In light of the above observations, TA modules could be essential genes for mycobacteria. At the same time their large number in the mycobacterial genome points towards possibilities of redundancy and pseudogene behaviour. Whole genome screens using transposon libraries have been conducted in M. tuberculosis to identify essential genes63,64. The screen includes several of the TA genes; however, none of them fall in to the category of essential genes. This is not surprising since knockout of all the ten TA loci in E. coli had no phenotype for normal growth and survival. Nonetheless deletion of multiple toxin genes drastically reduced tolerance to antibiotics and the level of persisters7 suggesting that TA genes provided selective survival advantage to cells under stress. Analysis of a recent essentiality screen data substantiates this observation65. Here, TA loci including higBA1, higBA2, parDE2, VapBC6, VapBC13 and UCAT3 are listed to confer growth advantage to mycobacteria. In our study also, higBA1, higBA2 and parDE2 are key TA modules. Essentiality of a gene for survival of the organism is dictated by the environment and a gene which is non-essential under normal growth conditions may become important under adverse conditions. TA loci appear to be one such set of genes which maybe non-essential under favourable growth conditions but confer survival advantage to the bacteria under unfavourable, stress conditions as are encountered in the human host.
Another important finding that emerges from this study is that even though the TA locus is an operon and its mRNA codes for both the toxin and antitoxin partner, the actual mRNA levels for the toxin and antitoxin genes are different. This has been reported in earlier publications32,36. Singh et al. have shown higher levels of toxin RNA compared to cognate antitoxin RNA for the three RelBE loci in lungs of M. tuberculosis-infected mice36. Korch et al. have also shown differences in levels of toxin and antitoxin RNA for the RelBE loci in M. tuberculosis when grown under hypoxic and starvation conditions32. The differential level of toxin and antitoxin mRNA in the cell at a given time could be a mechanism for regulating the ratio of antitoxin to toxin in the cell. Differential degradation of polycistronic RNA has been shown to determine differential expression of genes within an operon in several prokaryotic systems including E. coli, Salmonella typhimurium and Rhodobacter capsulatus66,67,68,69,70,71,72; however, the mechanisms involved are poorly understood. Degradation has been found to be RNaseE-dependent70, mRNA secondary structure based72,73 and small regulatory RNA (sRNA) induced74. Differential dosage of transcripts for kis and kid of the parD stability system of plasmid R1 has been shown to be a consequence of limited degradation of the polycistronic parD mRNA75. This plasmid addiction system is similar to the chromosomal toxin-antitoxin system. The expression of mycobacterial nucleases and proteases is also upregulated especially under STR and STRV stress. Protease-mediated degradation of antitoxins is well studied in E. coli and Streptococcus aureus48,49,50,51 and has been shown to contribute to increased toxin activity with enhanced drug resistance and persistence76. Similar studies in M. tuberculosis will shed light on the relevance of these observations and the role of nucleases and proteases in TA activation.
In summary, the present study provides vital information and clues for further studies on TA members in a genome by giving (i) direct experimental evidence for functionality of a system, (ii) configuration of the system in a genome, (iii) the activeness and responsiveness of a system to stimulus, (iv) a comprehensive analysis of the entire system across multiple time points and conditions, and (v) resolution of the system to identify key players with evidence for a division of labor across the TA system.
Methods
Growth of M. tuberculosis and RNA isolation
M. tuberculosis H37Rv was grown in Middlebrook 7H9 (Difco, Becton-Dickinson and Co., USA) supplemented with 0.05% Tween 80 and 10% albumin-dextrose-catalase (ADC, Difco, Becton-Dickinson and Co., USA) at 37 °C and 200 rpm, till an OD600 of 0.4–0.5. The culture was then split for addition of Isoniazid (isonicotinylhydrazine), Streptomycin and Ethambutol dissolved in autoclaved distilled water at a final concentration of 1 µg/ml and Rifampicin dissolved in ethanol at 0.1 µg/ml. Incubation was continued after addition for different time periods and control cultures (vehicle only) were also grown for same time periods. Cell density was measured as OD600 at all time points and CFU assay was performed. To prepare biological replicates for RNA isolation, each culture was independently grown in duplicate. Cells were harvested after 4, 6, 24, and 72 hrs of drug treatment and RNA was isolated. For nutrient starvation, the log phase culture of M. tuberculosis (OD600 of 0.4–0.5) was pelleted, washed twice with Phosphate Buffer Saline (PBS; 50 mM phosphate buffer, pH 7.4; 150 mM NaCl) containing 0.05% Tween 80, suspended in PBS and grown for 24 hrs and 1 week in tight-capped bottles as standing culture. Untreated culture grown in 7H9 served as control. For pH stress, the log phase culture of mycobacteria was re-suspended in Middlebrook 7H9 media with pH 5.2/pH 4.8 and grown for 24 and 72 hrs at 200 rpm. Control cultures were grown in 7H9 media with pH 6.8. The cells were harvested by centrifugation at 5000 g for 5 minutes at 4 °C and used for RNA isolation.
RNA isolation and Quantitative RT-PCR was performed using previously described methods77.
Microarray processing and data analysis
Using random nucleotide-based T7 promoter primers included in the Low Input Quick Amp WT labelling kit (Agilent Technologies, USA), 25 ng of RNA was amplified and labelled. The labelled cRNA were purified using RNeasy columns (QIAGEN Inc, USA). cRNA yields and specific activities were measured using Nanodrop 2000c spectrophotometer (Thermo Scientific, USA). Microarrays were synthesized using the proprietary non-contact SurePrint Ink-jet technology (Agilent Technologies, USA). The microarray specifications and design have been described earlier44. For all arrays, the Gene Expression Hybridization Kit (Agilent Technologies, USA) and recommended protocols were employed. Hybridization and slide processing were done according to the procedure described by Agilent Technologies. Microarray slides were scanned at a resolution of 5 µm using an Agilent Microarray scanner (G2565CA) with scan control software as per the manufacturer’s instructions. Raw data obtained were normalized and analyzed as described previously44. Probes that showed a 2-fold or higher change with a p value < 0.05 (unpaired Student t-test) were considered to be differentially expressed.
To identify differentially expressed genes that were unique to a given condition and those that were common with other conditions, a matrix was created using GeneMatrix software (Bionivid Technology Pvt Ltd, Bangalore, India). For this, the list of differentially expressed genes in each condition was provided as input to the software that computes all condition vs all condition matrix and identifies genes differentially expressed in one condition and those expressed across multiple conditions. TA genes 1.5 fold up and down regulated along with their fold change were subjected to unsupervised hierarchical clustering using Cluster 3.0 software by applying Pearson Uncentered algorithm with average linkage rule. The resultant cluster file was imported into Java Treeview software to visualize the cluster.
For determination of TA activation levels, the difference in fold change levels of toxin transcript level to antitoxin transcript level for all stress conditions and time points was calculated. If this median fold change difference increased from an early time point to a later time point under stress, it was designated as TA activation while if it decreased from an early to a later time point, it was designated as TA deactivation. If the difference remained equal to 1, it was designated co-expression. This model was used to calculate activation/deactivation scores and determine the pattern of TA response to a particular stress condition. Bi-clustering and Hierarchical clustering using Pearson uncentered algorithm with average linkage rule was used to identify the list of TA loci that were activated and deactivated. To construct the regulatory network for TA loci, the list of activated and deactivated TA loci along with the condition in which it was measured were provided as input to BridgeIsland software (Bionivid Technology Pvt Ltd, India). BridgeIsland software identifies the connecting nodes and edges from the raw input and derives the list of connections (TA– > Activation/Deactivation– > Condition) enriched in the overall experiment. Further, this information is provided as an input to CytoScape V 2.8.2 (NIGMS, NIH, USA) to visualize the network. Force-directed spring-embedded layout algorithm was applied to the network and nodes were sized based on their connectivity score with larger nodes bearing the highest score.
Change history
10 May 2018
A correction to this article has been published and is linked from the HTML and PDF version of this paper. The error has been fixed in the paper.
References
Gomez, J. E. & McKinney, J. D. M. tuberculosis persistence, latency, and drug tolerance. Tuberculosis (Edinb) 84, 29–44 (2004).
Stewart, G. R., Robertson, B. D. & Young, D. B. Tuberculosis: a problem with persistence. Nature reviews. Microbiology 1, 97–105, doi:10.1038/nrmicro749 (2003).
Christensen, S. K., Mikkelsen, M., Pedersen, K. & Gerdes, K. RelE, a global inhibitor of translation, is activated during nutritional stress. Proceedings of the National Academy of Sciences of the United States of America 98, 14328–14333 (2001).
Fu, Z., Tamber, S., Memmi, G., Donegan, N. P. & Cheung, A. L. Overexpression of MazFsa in Staphylococcus aureus induces bacteriostasis by selectively targeting mRNAs for cleavage. Journal of bacteriology 191, 2051–2059, doi:10.1128/JB.00907-08 (2009).
Gerdes, K., Christensen, S. K. & Lobner-Olesen, A. Prokaryotic toxin-antitoxin stress response loci. Nature reviews. Microbiology 3, 371–382, doi:10.1038/nrmicro1147 (2005).
Keren, I., Shah, D., Spoering, A., Kaldalu, N. & Lewis, K. Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. Journal of bacteriology 186, 8172–8180, doi:10.1128/JB.186.24.8172-8180.2004 (2004).
Maisonneuve, E., Shakespeare, L. J., Jorgensen, M. G. & Gerdes, K. Bacterial persistence by RNA endonucleases. Proceedings of the National Academy of Sciences of the United States of America 108, 13206–13211, doi:10.1073/pnas.1100186108 (2011).
Engelberg-Kulka, H. & Glaser, G. Addiction modules and programmed cell death and antideath in bacterial cultures. Annual review of microbiology 53, 43–70 (1999).
de Feyter, R., Wallace, C. & Lane, D. Autoregulation of the ccd operon in the F plasmid. Molecular & general genetics: MGG 218, 481–486 (1989).
Magnuson, R., Lehnherr, H., Mukhopadhyay, G. & Yarmolinsky, M. B. Autoregulation of the plasmid addiction operon of bacteriophage P1. The Journal of biological chemistry 271, 18705–18710 (1996).
Magnuson, R. & Yarmolinsky, M. B. Corepression of the P1 addiction operon by Phd and Doc. Journal of bacteriology 180, 6342–6351 (1998).
Bernard, P. & Couturier, M. Cell killing by the F plasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes. Journal of molecular biology 226, 735–745 (1992).
Christensen, S. K., Pedersen, K., Hansen, F. G. & Gerdes, K. Toxin-antitoxin loci as stress-response-elements: ChpAK/MazF and ChpBK cleave translated RNAs and are counteracted by tmRNA. Journal of molecular biology 332, 809–819 (2003).
Critchlow, S. E. et al. The interaction of the F plasmid killer protein, CcdB, with DNA gyrase: induction of DNA cleavage and blocking of transcription. Journal of molecular biology 273, 826–839 (1997).
Hurley, J. M., Cruz, J. W., Ouyang, M. & Woychik, N. A. Bacterial toxin RelE mediates frequent codon-independent mRNA cleavage from the 5′ end of coding regions in vivo. The Journal of biological chemistry 286, 14770–14778, doi:10.1074/jbc.M110.108969 (2011).
Hurley, J. M. & Woychik, N. A. Bacterial toxin HigB associates with ribosomes and mediates translation-dependent mRNA cleavage at A-rich sites. The Journal of biological chemistry 284, 18605–18613, doi:10.1074/jbc.M109.008763 (2009).
Lopes, A. P. et al. VapC from the leptospiral VapBC toxin-antitoxin module displays ribonuclease activity on the initiator tRNA. PloS one 9, e101678, doi:10.1371/journal.pone.0101678 (2014).
Munoz-Gomez, A. J., Santos-Sierra, S., Berzal-Herranz, A., Lemonnier, M. & Diaz-Orejas, R. Insights into the specificity of RNA cleavage by the Escherichia coli MazF toxin. FEBS letters 567, 316–320, doi:10.1016/j.febslet.2004.05.005 (2004).
Pedersen, K. et al. The bacterial toxin RelE displays codon-specific cleavage of mRNAs in the ribosomal A site. Cell 112, 131–140 (2003).
Gerdes, K. Toxin-antitoxin modules may regulate synthesis of macromolecules during nutritional stress. Journal of bacteriology 182, 561–572 (2000).
Lewis, K. Persister cells. Annual review of microbiology 64, 357–372 (2010).
Vazquez-Laslop, N., Lee, H. & Neyfakh, A. A. Increased persistence in Escherichia coli caused by controlled expression of toxins or other unrelated proteins. Journal of bacteriology 188, 3494–3497, doi:10.1128/JB.188.10.3494-3497.2006 (2006).
Amitai, S., Yassin, Y. & Engelberg-Kulka, H. MazF-mediated cell death in Escherichia coli: a point of no return. Journal of bacteriology 186, 8295–8300 (2004).
Jensen, R. B. & Gerdes, K. Programmed cell death in bacteria: proteic plasmid stabilization systems. Molecular microbiology 17, 205–210 (1995).
Yarmolinsky, M. B. Programmed cell death in bacterial populations. Science 267, 836–837 (1995).
Pandey, D. P. & Gerdes, K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic acids research 33, 966–976, doi:10.1093/nar/gki201 (2005).
Ramage, H. R., Connolly, L. E. & Cox, J. S. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS genetics 5, e1000767, doi:10.1371/journal.pgen.1000767 (2009).
Sala, A., Bordes, P. & Genevaux, P. Multiple toxin-antitoxin systems in Mycobacterium tuberculosis. Toxins 6, 1002–1020, doi:10.3390/toxins6031002 (2014).
Fivian-Hughes, A. S. & Davis, E. O. Analyzing the regulatory role of the HigA antitoxin within Mycobacterium tuberculosis. Journal of bacteriology 192, 4348–4356 (2010).
Gupta, A. Killing activity and rescue function of genome-wide toxin-antitoxin loci of Mycobacterium tuberculosis. FEMS microbiology letters 290, 45–53, doi:10.1111/j.1574-6968.2008.01400.x (2009).
Korch, S. B., Contreras, H. & Clark-Curtiss, J. E. Three Mycobacterium tuberculosis Rel toxin-antitoxin modules inhibit mycobacterial growth and are expressed in infected human macrophages. Journal of bacteriology 191, 1618–1630, doi:10.1128/JB.01318-08 (2009).
Korch, S. B., Malhotra, V., Contreras, H. & Clark-Curtiss, J. E. The Mycobacterium tuberculosis relBE toxin:antitoxin genes are stress-responsive modules that regulate growth through translation inhibition. J Microbiol 53, 783–795, doi:10.1007/s12275-015-5333-8 (2015).
Miallau, L. et al. Structure and proposed activity of a member of the VapBC family of toxin-antitoxin systems. VapBC-5 from Mycobacterium tuberculosis. The Journal of biological chemistry 284, 276–283 (2009).
Schifano, J. M. et al. Mycobacterial toxin MazF-mt6 inhibits translation through cleavage of 23S rRNA at the ribosomal A site. Proceedings of the National Academy of Sciences of the United States of America 110, 8501–8506, doi:10.1073/pnas.1222031110 (2013).
Sharp, J. D. et al. Growth and translation inhibition through sequence-specific RNA binding by Mycobacterium tuberculosis VapC toxin. The Journal of biological chemistry 287, 12835–12847, doi:10.1074/jbc.M112.340109 (2012).
Singh, R., Barry, C. E. 3rd & Boshoff, H. I. The three RelE homologs of Mycobacterium tuberculosis have individual, drug-specific effects on bacterial antibiotic tolerance. Journal of bacteriology 192, 1279–1291, doi:10.1128/JB.01285-09 (2010).
Yang, M., Gao, C., Wang, Y., Zhang, H. & He, Z. G. Characterization of the interaction and cross-regulation of three Mycobacterium tuberculosis RelBE modules. PloS one 5, e10672 (2010).
Zhu, L. et al. The mRNA interferases, MazF-mt3 and MazF-mt7 from Mycobacterium tuberculosis target unique pentad sequences in single-stranded RNA. Molecular microbiology 69, 559–569, doi:10.1111/j.1365-2958.2008.06284.x (2008).
Zhu, L. et al. Characterization of mRNA interferases from Mycobacterium tuberculosis. The Journal of biological chemistry 281, 18638–18643, doi:10.1074/jbc.M512693200 (2006).
Dahl, J. L. et al. The role of RelMtb-mediated adaptation to stationary phase in long-term persistence of Mycobacterium tuberculosis in mice. Proceedings of the National Academy of Sciences of the United States of America 100, 10026–10031, doi:10.1073/pnas.1631248100 (2003).
Demidenok, O. I., Kaprelyants, A. S. & Goncharenko, A. V. Toxin-antitoxin vapBC locus participates in formation of the dormant state in Mycobacterium smegmatis. FEMS microbiology letters 352, 69–77, doi:10.1111/1574-6968.12380 (2014).
Tiwari, P. et al. MazF ribonucleases promote Mycobacterium tuberculosis drug tolerance and virulence in guinea pigs. Nature communications 6, 6059, doi:10.1038/ncomms7059 (2015).
Ahidjo, B. A. et al. VapC toxins from Mycobacterium tuberculosis are ribonucleases that differentially inhibit growth and are neutralized by cognate VapB antitoxins. PloS one 6, e21738, doi:10.1371/journal.pone.0021738 (2011).
Venkataraman, B., Vasudevan, M. & Gupta, A. A new microarray platform for whole-genome expression profiling of Mycobacterium tuberculosis. Journal of microbiological methods 97, 34–43, doi:10.1016/j.mimet.2013.12.009 (2014).
Boshoff, H. I. et al. The transcriptional responses of Mycobacterium tuberculosis to inhibitors of metabolism: novel insights into drug mechanisms of action. The Journal of biological chemistry 279, 40174–40184, doi:10.1074/jbc.M406796200 (2004).
Morris, R. P. et al. Ancestral antibiotic resistance in Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences of the United States of America 102, 12200–12205, doi:10.1073/pnas.0505446102 (2005).
Betts, J. C., Lukey, P. T., Robb, L. C., McAdam, R. A. & Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Molecular microbiology 43, 717–731 (2002).
Brzozowska, I. & Zielenkiewicz, U. The ClpXP protease is responsible for the degradation of the Epsilon antidote to the Zeta toxin of the streptococcal pSM19035 plasmid. The Journal of biological chemistry 289, 7514–7523, doi:10.1074/jbc.M113.519488 (2014).
Christensen, S. K. et al. Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: involvement of the yefM-yoeB toxin-antitoxin system. Molecular microbiology 51, 1705–1717 (2004).
Donegan, N. P., Thompson, E. T., Fu, Z. & Cheung, A. L. Proteolytic regulation of toxin-antitoxin systems by ClpPC in Staphylococcus aureus. Journal of bacteriology 192, 1416–1422, doi:10.1128/JB.00233-09 (2010).
Van Melderen, L., Bernard, P. & Couturier, M. Lon-dependent proteolysis of CcdA is the key control for activation of CcdB in plasmid-free segregant bacteria. Molecular microbiology 11, 1151–1157 (1994).
Carroll, P., Brown, A. C., Hartridge, A. R. & Parish, T. Expression of Mycobacterium tuberculosis Rv1991c using an arabinose-inducible promoter demonstrates its role as a toxin. FEMS microbiology letters 274, 73–82, doi:10.1111/j.1574-6968.2007.00842.x (2007).
Han, J. S. et al. Characterization of a chromosomal toxin-antitoxin, Rv1102c-Rv1103c system in Mycobacterium tuberculosis. Biochemical and biophysical research communications 400, 293–298, doi:10.1016/j.bbrc.2010.08.023 (2010).
Huang, F. & He, Z. G. Characterization of an interplay between a Mycobacterium tuberculosis MazF homolog, Rv1495 and its sole DNA topoisomerase I. Nucleic acids research 38, 8219–8230, doi:10.1093/nar/gkq737 (2010).
Robson, J., McKenzie, J. L., Cursons, R., Cook, G. M. & Arcus, V. L. The vapBC operon from Mycobacterium smegmatis is an autoregulated toxin-antitoxin module that controls growth via inhibition of translation. Journal of molecular biology 390, 353–367, doi:10.1016/j.jmb.2009.05.006 (2009).
Schuessler, D. L. et al. Induced ectopic expression of HigB toxin in Mycobacterium tuberculosis results in growth inhibition, reduced abundance of a subset of mRNAs and cleavage of tmRNA. Molecular microbiology 90, 195–207, doi:10.1111/mmi.12358 (2013).
Zhao, L. & Zhang, J. Biochemical characterization of a chromosomal toxin-antitoxin system in Mycobacterium tuberculosis. FEBS letters 582, 710–714, doi:10.1016/j.febslet.2008.01.045 (2008).
Frampton, R., Aggio, R. B., Villas-Boas, S. G., Arcus, V. L. & Cook, G. M. Toxin-antitoxin systems of Mycobacterium smegmatis are essential for cell survival. The Journal of biological chemistry 287, 5340–5356, doi:10.1074/jbc.M111.286856 (2012).
Albrethsen, J. et al. Proteomic profiling of Mycobacterium tuberculosis identifies nutrient-starvation-responsive toxin-antitoxin systems. Molecular & cellular proteomics: MCP 12, 1180–1191, doi:10.1074/mcp.M112.018846 (2013).
McKenzie, J. L. et al. A VapBC toxin-antitoxin module is a posttranscriptional regulator of metabolic flux in mycobacteria. Journal of bacteriology 194, 2189–2204, doi:10.1128/JB.06790-11 (2012).
Keren, I., Minami, S., Rubin, E. & Lewis, K. Characterization and transcriptome analysis of Mycobacterium tuberculosis persisters. mBio 2, e00100–00111, doi:10.1128/mBio.00100-11 (2011).
Zhang, Y. et al. MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Molecular cell 12, 913–923 (2003).
Griffin, J. E. et al. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS pathogens 7, e1002251, doi:10.1371/journal.ppat.1002251 (2011).
Zhang, Y. J. et al. Global assessment of genomic regions required for growth in Mycobacterium tuberculosis. PLoS pathogens 8, e1002946, doi:10.1371/journal.ppat.1002946 (2012).
DeJesus, M. A. et al. Comprehensive Essentiality Analysis of the Mycobacterium tuberculosis Genome via Saturating Transposon Mutagenesis. mBio 8, doi:10.1128/mBio.02133-16 (2017).
Baga, M., Goransson, M., Normark, S. & Uhlin, B. E. Processed mRNA with differential stability in the regulation of E. coli pilin gene expression. Cell 52, 197–206 (1988).
Belasco, J. G., Beatty, J. T., Adams, C. W., von Gabain, A. & Cohen, S. N. Differential expression of photosynthesis genes in R. capsulata results from segmental differences in stability within the polycistronic rxcA transcript. Cell 40, 171–181 (1985).
McCarthy, J. E. Post-transcriptional control in the polycistronic operon environment: studies of the atp operon of Escherichia coli. Molecular microbiology 4, 1233–1240 (1990).
Newbury, S. F., Smith, N. H., Robinson, E. C., Hiles, I. D. & Higgins, C. F. Stabilization of translationally active mRNA by prokaryotic REP sequences. Cell 48, 297–310 (1987).
Nilsson, P. & Uhlin, B. E. Differential decay of a polycistronic Escherichia coli transcript is initiated by RNaseE-dependent endonucleolytic processing. Molecular microbiology 5, 1791–1799 (1991).
Owolabi, J. B. & Rosen, B. P. Differential mRNA stability controls relative gene expression within the plasmid-encoded arsenical resistance operon. Journal of bacteriology 172, 2367–2371 (1990).
Yu, X., Zheng, W., Bhat, S., Aquilina, J. A. & Zhang, R. Transcriptional and posttranscriptional regulation of Bacillus sp. CDB3 arsenic-resistance operon ars1. PeerJ 3, e1230, doi:10.7717/peerj.1230 (2015).
Newbury, S. F., Smith, N. H. & Higgins, C. F. Differential mRNA stability controls relative gene expression within a polycistronic operon. Cell 51, 1131–1143 (1987).
Desnoyers, G., Morissette, A., Prevost, K. & Masse, E. Small RNA-induced differential degradation of the polycistronic mRNA iscRSUA. The EMBO journal 28, 1551–1561, doi:10.1038/emboj.2009.116 (2009).
Ruiz-Echevarria, M. J., de la Cueva, G. & Diaz-Orejas, R. Translational coupling and limited degradation of a polycistronic messenger modulate differential gene expression in the parD stability system of plasmid R1. Molecular & general genetics: MGG 248, 599–609 (1995).
Baek, K. T. et al. beta-Lactam resistance in methicillin-resistant Staphylococcus aureus USA300 is increased by inactivation of the ClpXP protease. Antimicrobial agents and chemotherapy 58, 4593–4603, doi:10.1128/AAC.02802-14 (2014).
Venkataraman, B., Gupta, N. & Gupta, A. A robust and efficient method for the isolation of DNA-free, pure and intact RNA from Mycobacterium tuberculosis. Journal of microbiological methods 93, 198–202, doi:10.1016/j.mimet.2013.03.018 (2013).
Acknowledgements
This work was financially supported by Department of Biotechnology, Govt. of India. Processing of M. tuberculosis cultures was carried out in the BSL-3 facility at UDSC. Microarray experiments were performed at DBT-supported Genomic Facility at University of Delhi South Campus. Dr. V. K. Chaudhary and Dr. Vandana Malhotra are acknowledged for critical reading of the manuscript.
Author information
Authors and Affiliations
Contributions
A.G. conceived study, planned experiments, analyzed data and wrote the manuscript; V.B. grew M. tuberculosis cultures, isolated RNA and performed the microarray experiments; M.V. and K.B. performed bioinformatics analysis and representation of data for the manuscript in discussion with A.G.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gupta, A., Venkataraman, B., Vasudevan, M. et al. Co-expression network analysis of toxin-antitoxin loci in Mycobacterium tuberculosis reveals key modulators of cellular stress. Sci Rep 7, 5868 (2017). https://doi.org/10.1038/s41598-017-06003-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-06003-7
This article is cited by
-
Are all VapC toxins of Mycobacterium tuberculosis endowed with enigmatic RNase activity?
Journal of Biosciences (2024)
-
Revolutionizing control strategies against Mycobacterium tuberculosis infection through selected targeting of lipid metabolism
Cellular and Molecular Life Sciences (2023)
-
Functional characterization of HigBA toxin-antitoxin system in an Arctic bacterium, Bosea sp. PAMC 26642
Journal of Microbiology (2022)
-
Modulatory Impact of the sRNA Mcr11 in Two Clinical Isolates of Mycobacterium tuberculosis
Current Microbiology (2022)
-
A YoeB toxin cleaves both RNA and DNA
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
