
Abstract
Epinephelus awoara, as known as yellow grouper, is a significant economic marine fish that has been bred artificially in China. However, the genetic structure and evolutionary history of yellow grouper remains largely unknown. Here, this work presents the high-quality chromosome-level genome assembly of yellow grouper using PacBio single molecule sequencing technique (SMRT) and High-through chromosome conformation capture (Hi-C) technologies. The 984.48 Mb chromosome-level genome of yellow grouper was assembled, with a contig N50 length of 39.77 Mb and scaffold N50 length of 41.39 Mb. Approximately 99.76% of assembled sequences were anchored into 24 pseudo-chromosomes with the assistance of Hi-C reads. Furthermore, approximately 41.17% of the genome was composed of repetitive elements. In total, 24,541 protein-coding genes were predicted, of which 22,509 (91.72%) genes were functionally annotated. The highly accurate, chromosome-level reference genome assembly and annotation are crucial to the understanding of population genetic structure, adaptive evolution and speciation of the yellow grouper.
Similar content being viewed by others


Background & Summary
Groupers belongs to the subfamily Epinephelinae under the family Epinephelidae, which mainly inhabits tropical and subtropical coral reefs or continental shelves, acting as the top predators maintaining the ecological balance of coral reef ecosystems1. Groupers encompasses over 16 genera and more than 160 species, out of which approximately 47 species were currently cultivated for aquaculture2, making them globally significant economic fish species. According to statistics from the Food and Agriculture Organization (FAO), the global aquaculture production of groupers in 2020 amounted to 226.2 thousand tonnes3. In China, as per data from the China Fisheries Statistical Yearbook 2023, the aquaculture production of groupers in 2022 reached 205.8 thousand tonnes, ranking it fourth in terms of marine aquaculture production in China4. In conclusion, groupers hold significant ecological aimportance and commercial value. In recent years, several high-quality chromosome-level assemblies of grouper reference genomes have been assembled, including seven species of the Epinephelus genus5,6,7,8,9,10,11,12,13, one species of the Plectropomus genus14,15,16,17, one species of the Cromileptes genus18, and one species of the Cephalopholis genus19, which provide important genomic resources for evolutionary analysis, molecular-assisted breeding, and germplasm conservation of groupers. Phylogenetic studies show that Epinephelus species in the western Pacific Ocean have diverged into two genetic clades with variations in body size20,21, implying the potential presence of independent evolutionary trajectories during the course of adaptation. However, a significant limitation exists, as reference genomes are available for seven species within the large-bodied clade5,7,8,9,10,11,12,13,18, whereas the small-bodied clade has an accessible reference genome for only one specie, the E. akaara6. This limitation hampers research on the body size differentiation, speciation, and evolutionary history of the groupers.
The subject of this study, E. awoara, commonly referred to as the yellow grouper, is widely distributed in occurs in the western North Pacific Ocean. Its distribution spans regions such as North and South Korea, Japan, China, Vietnam, and islands in the South China Sea, with occasional occurrences in the eastern Indian Ocean22. Notably, yellow grouper is one of the few grouper species capable of inhabiting higher latitudes, approximately around the 40th parallel north (40 °N). It is worth noting that the yellow grouper is a protogynous hermaphroditic fish23, belongs to the small-sized clade of Epinephelus species in the western Pacific Ocean20,21. It serves as valuable material for exploring sex change and body size differentiation in groupers. Distinguished by its striking appearance, characterized by a golden-yellow color on its abdomen, with five distinct banding patterns and prominently gilded outlines on the dorsal and caudal fins (Fig. 2b), yellow grouper has gained recognition and popularity among consumers and aquaculturists due to its delectable taste and considerable economic value. Presently, this species has been successfully subjected to artificial aquaculture in China, and has been utilized in hybrid breeding programs24. Consequently, it is considered as an economically promising fish species in the market. Despite broad research interests in this species, such as reproductive biology23, artificial breeding24,25, cytogenetics26, genetic structure27,28, and phylogenetics29, studies related to its genome-based breeding, evolution, and germplasm resources have been limited. Notably, only one report on the complete mitochondrial (mt) genome was available has delved into genetic resources29. Therefore, the assembly of a high-quality genome for yellow grouper is of paramount significance, and would provide an essential genome resource for molecular genetics, molecular breeding, evolutionary studies, and germplasm conservation of yellow grouper.
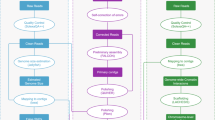
The third-generation sequencing technology can produce genomes with high integrity through single-molecule real-time (SMRT) sequencing technology and high-throughput chromatin conformation capture (Hi-C) technology can achieve genome assembly at the chromosome level14,30,31,32. Recently, Pacific Biosciences released a new highly accurate long-read sequencer called the Revio System. In this study, we have employed a combination of PacBio Revio long-read circular consensus sequence (ccs) data and Hi-C technology to generate a high-quality, chromosome-level assembly of the yellow grouper genome. The workflow of de novo genome assembly and annotation is shown in the Fig. 1. The highly accurate, chromosome-level reference genome is essential to support basic genetic studies and will be contribute to genetic structure, evolutionary studies and germplasm conservation for yellow grouper.

The overview of the chromosome-level genome assembly and annotation. Chrs: chromosomes. We first used 56.81 Gb short-read sequencing data to predict the assembled genome size was approximately 990.21 Mb Gb by K-mer analysis, and the GC content and heterozygosity were approximately 41.47% and 0.40%, respectively. Then, the 43.32 Gb of PacBio ccs data resulted in a 984.48 Mb assembly covers 98.60% of the genome, with contig N50 of 40.27 Mb. The 40 contigs were anchored into 24 pseudo-chromosomes covering roughly 99.76% of the genome assembly with the assistance of 114.71 Gb Hi-C reads. The final assembly consisted of 24 pseudo-chromosomes that yielded 984.48 Mb of yellow grouper genome, with a contig N50 of 39.77 Mb and scafold N50 of 43.39 Mb. The genome contained 35.96% repeat sequences and 8,235 noncoding RNAs. Additionally, 22,509 (91.72%) genes were functionally annotated from a total of 24,541 predicted protein-coding genes by combination of RNAseq and ISO-Seq annotation, genome sequence, and homolog protein.
Methods
Ethics statement
All experiments were performed according to the Guidelines for the Care and Use of Laboratory Animals in China. All experimental procedures and sample collection methods were approved by the Institutional Animal Care and Use Committee (IACUC) of the School of Life Sciences, Sun Yat-sen University under approval No. SYSU-IACUS-2022-B0129.
Sample collection, library construction and sequencing
A wild female yellow grouper (body mass: 221.0 g) was collected at coastal of Daya Bay, Huizhou, Guangdong (22°43′1.28″ N; 114°32′14.54″ E). However, in fact, under natural conditions, yellow grouper are protogynous hermaphrodites 23. The muscle was collected for DNA extraction and was used for short-read sequencing, long-read SMRT sequencing, and Hi-C sequencing. 11 different tissues (muscle, gill, skin, liver, intestine, spleen, kidney, head-kidney, heart, eye, and brain) were collected for RNA extraction, then mixed into a total RNA for RNA Sequencing. All samples were immediately frozen in liquid nitrogen and then stored at −80 °C.
Library construction and sequencing
All the DNA extraction, library construction and sequencing procedures were performed by the Nextomics Biosciences (Wuhan, China) according to the manufacturer’s protocols. Briefly, high molecular weight genomic DNA was extracted from muscle for muscle for MGI sequencing and PacBio SMRT sequencing. The DNA quality and purity of the extracted DNA was monitored on 0.75% agarose gels and NanoDrop One UV-Vis spectrophotometer (Thermo Fisher Scientific, USA). At last, DNA concentration of 316.0 ng/µL was measured by Qubit Fluorometer (Invitrogen, USA). High-quality DNA was used for library preparation and high-throughput sequencing.
For short-read sequencing, the genomic DNA (gDNA) was randomly fragmented and then the library was prepared following the manufacturer’s instructions. The qualified libraries were sequenced on MGISEQ-2000 platform. A total of 56.71 Gb raw reads were generated for genome survey. The Q20 rate and Q30 rate were 98.39% and 94.60%, respectively, with an average coverage depth of 57.27× (Table 1).
For PacBio sequencing, the gDNA was used to construct SMRTbell libraries according to PacBio’s standard protocol (Pacific Biosciences, CA, USA) using 20 kb preparation solutions33. The SMRTbell library construction, inculding the DNA shearing, repair ends, and ligation of DNA fragments with hairpin adapters to create circular templates for SMRT sequencing. The library was sequenced using the PacBio Revio System in circular consensus sequence (CCS) mode following the manufacturer’s instructions. The quality control of the raw data was performed using ccs software (https://github.com/PacificBiosciences/ccs) with parameter of “-min-passes 1-min-rq 0.99-min-length 100”. As a result, 1 SMRT cell was sequenced, and a total of 43.32 Gb PacBio CCS reads (coverage depth as 43.75×) were generated for the subsequent genome assembly. The average read length and N50 length of the subreads sequences were 21,905 bp and 23,217 bp, respectively (Table 1).
For the Hi-C sequencing, the fresh muscle sample was fixed with 2% formaldehyde to produce crosslinking of the DNA-protein. The Hi-C library construction process involves crosslink DNA, digestion, biotin labelling, proximity ligation, and DNA purification, was prepared following the protocol34. Finally, the Hi-C library was subjected to paired-end sequencing with 150 bp read lengths using the MGISEQ-2000 platform to capture the spatial interactions between chromosomal regions. As a result, 109.97 Gb of Hi-C read data was generated, with an average sequencing coverage of 111.71× (Table 1).
RNA sequencing (RNA-Seq) using short-read sequencing technology was widely used method for transcriptome profiling35. While emerging single molecule, long-read RNA-Seq technologies have enabled new approaches to study the transcriptome and its function36. SMRT isoform sequencing (Iso-Seq) with the PacBio platform can generate full-length cDNA sequences37. Read lengths achieved with these technologies (~15 kb) surpass lengths of most transcripts. In this study, for substantiating transcripts to annotate the genome structure, we performed RNA-Seq and Iso-Seq of the total RNA, respectively. Total RNA was extracted by grinding tissue in TRIzol reagent (Tiangen) on dry ice and processed following the protocol provided by the manufacturer. The integrity of the RNA was determined with the Agilent 2100 Bioanalyzer (Agilent Technologies) and agarose gel electrophoresis. The purity and concentration of the RNA were determined with the Nanodrop (Thermo Fisher Scientific) and Qubit (Thermo Fisher Scientific). Then, equal amount of them were pooled together for RNA sequencing. Finally, sequencing of RNA-Seq and Iso-Seq were performed on the MGISEQ-2000 platform and the PacBio Sequel II platform, respectively. A total of 17.79 Gb RNA-seq data and 64.59 Gb clean Iso-Seq data were generated (Table 1), which were then used for whole-genome protein-coding gene prediction.
Genome survey
The k-mer analysis was performed using MGI paired-ended sequenced raw reads prior to genome assembly to estimate the genome size and heterozygosity. Briefly, 56.71 Gb raw dara was filtered by fastp v 0.21.038 software with parameters of “-n 0 -f 5 -F 5 -t 5 -T 5 -q 20”, and 52.65 Gb clean data were retained (Table 1). The quality-filtered clean reads were subjected to k-17mers frequency distribution and heterozygosity using the KMC program39 with parameters of “-k17 -ci1 -cs1000000”. The genome size was estimated using FindGSE software40 and GenomeScope (v 1.0.0)41 with parameters of “default”. Finally, a total of 34,657,425,513 k-mers were counted with a k-mers peak at a depth of 35 (Table 2). We estimated that the genome size of the yellow grouper = K-mer num/K-mer depth = 990.21 Mb. The heterozygosity rate was estimated to be approximately 0.40% on k-mer depth distribution (Table 2).
De novo assembly of the yellow grouper genome
The raw PacBio CCS reads data was used for de novo genome assembly using hifiasm v 0.19.442 with default parameters. To further improve the accuracy of the assembly, the preliminary assembled genome was polished by short reads from the same individual using four iterative correction rounds of Nextpolish (v1.2.443) with default parameters. To evaluate the accuracy of the assembly, all the Illumina paired end reads were mapped to the assembled genome using BWA (Burrows-Wheeler Aligner, v 0.7.12-r103944) and the mapping rate as well as genome coverage of sequencing reads were assessed using Minimap2 v r4145 with parameters of “-x map-pb”. Besides, base accuracy of the assembly was calculated with samtools v 1.446 and Bcftools v1.8.047 with default parameters. To avoid including mitochondria sequences in the assembly, the draft genome assembly was submitted to the NT library and aligned sequences were eliminated using the blast v2.948. The resulting assembly consists of 64 contigs and has a total length of 984.53 Mb with a contig N50 length of 40.27 Mb (Table 3).
Pseudochromosome construction
In total, 109.97 Gb clean paired-end reads were generated from the libraries. Firstly, low-quality sequences (quality scores <20), adaptor sequences and sequences shorter than 30 bp were filtered out using fastp v0.21.038 with default parameters. Then, the clean paired-end reads were mapped to the draft assembled sequence using bowtie2 v 2.3.249 with parameters of “-end-to-end,–very-sensitive -L 30” to get the unique mapped paired-end reads. Valid interaction paired reads (invalid read pairs, including dangling-end, self-cycle, re-ligation, and dumped products were filtered) were identified and retained by HiC-Pro v 3.1.050 from unique mapped paired-end reads for further analysis. The scaffolds were further clustered, ordered, and oriented scaffolds onto chromosomes by Lachesis51 with parameters of CLUSTER MIN RE SITES = 100, CLUSTER MAX LINK DENSITY = 2.5, CLUSTER NONINFORMATIVE RATIO = 1.4, ORDER MIN N RES IN TRUNK = 60, and ORDER MIN N RES IN SHREDS = 60. Finally, placement and orientation errors exhibiting obvious discrete chromatin interaction patterns were manually adjusted. Following the scaffolding procedure, the 974.86 Mb were successfully anchored to the 24 chromosomes with an integration efficiency of 99.02%, and the lengths of chromosomes ranged from 23.08 Mb to 48.78 Mb (Table 4). After Hi-C scaffolding, the 984.48 Mb chromosome-level genome of yellow grouper was assembled, with a contig N50 length of 39.77 Mb and a scaffold N50 length of 41.39 Mb (Table 3). Moreover, we evaluated the result of Hi-C based pseudo-chromosomes construction. The 24 scaffolds are clearly distinguishable in the heatmap, the interaction signal around the diagonal is strongly apparent (Fig. 2a), indicating the high-quality of the pseudochromosomes assembly.

Chromosome-level genome assembly and annotation of the yellow grouper. (a) Hi-C interaction heat map (100-kb resolution) for the assembled genome of yellow grouper. Abscissa and ordinate represent order of each bin on corresponding chromosome group. Color block illuminates intensity of interaction from yellow (low) to red (high). (b) Genomic landscape of yellow grouper. All represented in 50-kb genomic windows. (c) The genome completeness and QV was assessed using Merqury. (d) CEGMA assesses the assembly status of core genes within the assembled genome. (e) BUSCO evaluation on the genome assembly completeness of the assembled genome. (f) Venn diagram of function annotations from various databases. The Venn diagram displays the overlap and uniqueness of functional gene annotations derived from five databases: SwissProt, Non-Reduntant Protein Database (NR), Kyoto Encyclopedia of Gene and Genomes (KEGG), Eukaryotic Orthologous Groups of protein (KOG) and Gene Ontology (GO). (g) BUSCO evaluation of gene annotation in yellow grouper.
Repeat annotation
We first annotation the tandem repeats, including simple repeat sequences (SSRs) and tandem repeat elements, were identified using the software GMATA v2.252 and Tandem Repeats Finder (TRF V 4.07b53) with default parameters. Then, transposable elements (TE) in the yellow grouper genome were identified using a combination of ab initio and homology-based methods. Briefly, an ab initio repeat library was first predicted using MITE-hunter54 with parameters of “-n 20 -P 0.2 -c 3” and RepeatModeler version open-2.0.455 with parameters of “-engine wublast”, in which LTR_FINDER56, LTRharverst57 and LTR_retriver58 synchronously to detect repeat sequences in the yellow grouper genome. The obtainted library was then aligned to TEclass Repbase (http://www.girinst.org/repbase) to classify the type of each repeat family using TEclass v 2.1.359. For further identification of the repeats throughout the genome, RepeatMasker (open-4.1.4)60 was applied to search for known and novel TEs by mapping sequences against the de novo repeat library and Repbase TE library with parameters of “nolow -no_is -gff -norna -engine abblast -lib lib”. Overlapping transposable elements belonging to the same repeat class were collated and combined. A total of 405.30 Mb sequences, 41.17% of the yellow grouper genome, were identified as repeat elements (Table 5 and Fig. 2b). We estimated that the yellow grouper genome consists of approximately 0.73% tandem repeats, including 0.18% of SSR and 0.56% of tandem repeats in the genome (Table 5 and Fig. 2b). A significant portion of the genome, approximately 35.68%, is masked, resulting in 351.25 Mb being identified as transposable elements (Table 5 and Fig. 2b). Among these transposable elements, DNA transposons were the main type, occupying 20.08% (197.69 Mb) of the genome. Retroelements, including long interspersed nuclear elements (LINEs, 7.52%), long terminal repeats (LTRs, 4.24%), and short interspersed nuclear elements (SINEs, 1.02%), resulting the proportion of 12.78% of the genome (Table 5).
Gene model prediction and functional annotations
We conducted protein-coding gene prediction with three independent approaches, including homolog protein, transcriptome-assisted annotation, and de novo prediction, were used for gene prediction in a repeat-masked genome. For homology-based gene prediction, we utilized GeMoMa v1.6.161 with default parameters to align the protein-coding sequences frome E. fuscoguttatus (brown-marbled grouper9), E. moara (kelp grouper10), E. lanceolatus (giant grouper11), Cromileptes altivelis (humpback grouper18), Plectropomus leopardus (leopard coral grouper16), Danio rerio (zebrafish, GCF_000002035.662), and Oryzias latipes (Japanese medaka, GCF_002234675.1) to the genome assembly, and then got the gene structure information. For transcriptome-based prediction, the filtered long read Iso-seq and short-read RNA-seq data were aligned to the reference genome using STAR v2.7.3a63, followed by the transcripts were then assembled using Stringtie v1.3.4d64 and open reading frames (ORFs) were predicted using PASA v2.3.365 to produce a training set. For the de novo prediction, Augustus v3.3.166 with parameters of “--gff3 = on --hintsfile = hints.gff --extrinsicCfgFile = extrinsic.cfg --allow_hinted_splicesites = gcag,atac–min_intron_len = 30 --softmasking = 1” were then utilized for ab initio gene prediction with the training set. Finally, EVidenceModeler (EVM, v1.165) was used to produce an integrated gene set of which gene with TE were removed using TransposonPSI package (http://transposonpsi.sourceforge.net/) and the miscoded genes were further filtered. Untranslated regions (UTRs) and alternative splicing regions were determined using PASA v2.3.365 based on RNA-seq assemblies. We retained the longest transcripts for each locus, and regions outside of the ORFs were designated UTRs. Furthermore, we performed functional annotation of the predicted protein-coding genes via assigning by comparing with public databases including SwissProt67, the NCBI non-reduntant protein database (NR), Kyoto Encyclopedia of Gene and Genomes (KEGG)68, Eukaryotic Orthologous Groups of protein (KOG)69, and Gene Ontology (GO)70. The putative domains and GO terms of genes were identified using the InterProScan program with default parameters. For the other four databases, BLASTp (https://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to compare the EVidenceModeler-integrated protein sequences against the four well-known public protein database with parameters of “-evalue 1e-5, -max_target_seqs. 1”65. Results from the five database searches were concatenated using EVidenceModeler v1.165.
A total of 24,541 protein-coding genes were successfully predicted within the genome, with an average gene length and an average CDS length of 20,681.6 bp and 1,743.35 bp in each gene, respectively. The average exons number of 10.22, average exon length of 170.5 bp and average intron length of 2,052.99 in each gene (Table 6 and Fig. 2b). Further, 22,509 genes were successfully annotated, accounting for 91.72% of all predicted genes (Table 7 and Fig. 2f).
To obtain the non-coding RNA (ncRNA), two strategies were used: searching against database and prediction with model. Transfer RNAs (tRNAs) were predicted using tRNAscan-SE v2.071 with parameters “–thread 4 -E -I”. Micro RNA (miRNA), rRNA, small nuclear RNA, and small nucleolar RNA were detected using “cmscan” subprogram from Infernal v1.1.272 to search the Rfam database73 with following parameter. The rRNAs and their subunits were predicted using RNAmmer v1.274 with parameters “-S euk -m lsu,ssu,tsu -gff”. As a result, we annotated 1,295 rRNA, 1,946 miRNA, 276 regulatory and 2,391 tRNA (Table 8 and Fig. 2b).
Data Records
The raw sequence data, including the PacBio long-read data, MGI short-read genomic sequencing data, Hi-C data and Transcriptomic sequences, (including RNA-Seq and Iso-Seq data), have been deposited in the Genome Sequence Archive (GSA75) in National Genomics Data Center76 under the accession CRA01309777. Additionally, the raw data has also been deposited at NCBI with the accession number SRP47989378. The assembled genome sequences have been deposited in the NCBI GenBank with the accession number GCA_035609425.179. The whole genome sequence data and the genome annotation files reported in this paper have been deposited in the Genome Warehouse in National Genomics Data Center76,80, Beijing Institute of Genomics, Chinese Academy of Sciences / China National Center for Bioinformation, under accession number GWHEQBJ0000000081.
Technical Validation
Assembly completeness and accuracy were evaluated by multiple methods. First, the MGI short-read clean reads and PacBio long-read data (Table 1), were re-mapped onto the assembly using BWA v 0.7.12-r103944 and minimap245, respectively. The coverage rate of MGI WGS short-read and PacBio CCS long-read reached 98.87% and 97.66% of the assembly have at least 20× coverage, respectively (Table 9), demonstrating a high level of assembly accuracy. Then, the Merqury v1.382 was used to assess the genome quality, with consensus quality value (QV) and completeness statistic values of 52.10 and 91.84%, respectively, indicating a high level of accuracy and completeness in the assembled genome (Fig. 2c). The CEGMA v383 was employed to assess the accuracy and completeness of core genes within the assembled genome. A total of 243 core genes were assembled, accounting for 97.98% of the expected core genes. Among these, 194 were fully assembled, representing 78.23% completeness, indicating a relatively comprehensive representation of core genes in the assembled genome (Fig. 2d). Benchmarking Universal Single-Copy Orthologues (BUSCO) software v5.3.184 also used to evaluate the completeness of the assembly with parameters “-l actinopterygii_odb10 -g genome”. We identified 3589 complete BUSCOs (98.60%) out of the 3640 BUSCO groups, including 3536 complete and single-copy BUSCOs (97.14%) and 53 complete and duplicated BUSCOs (1.46%). The number of fragmented BUSCOs and missing BUSCOs was 16 (0.44%) and 35 (0.96%), respectively (Fig. 2e).
Furthermore, the completeness of gene annotations were evaluated using BUSCO v5.3.184 with the actinopterygii_odb10 database. The annotated genes covered a total of 96.87% (3526) of the complete vertebrate core gene set, indicating a high reliable of the gene prediction results (Fig. 2g). RNA-Seq reads were mapped with the annotation results, we used Stringtie v1.3.4d64 with default parameters and achieved an overall mapping rate of 91.76%. Next, we compared the number of genes, gene length, coding DNA sequence (CDS) length, exons number per gene, exon length, and intron length with those of other teleost fish species (Table 10).
Genome collinearity analysis and visualizations were performed using the MCScan tool from jcvi v1.3.885, obtained from https://github.com/tanghaibao/jcvi/wiki/MCscan-(Python-version). We illustrated the collinearity between the yellow grouper genome and other grouper species using collinearity plots. The yellow grouper genome demonstrates strong collinearity with related species within its genus and with the humpback grouper (C. altivelis) from a distinct genus (Fig. 3a,b). However, compared to another genus, the leopard coral grouper (P. leopardus), it exhibits more frequently chromosomes are rearranged. (Fig. 3b).

Genome collinearity between the yellow grouper and other grouper species. (a) Collinearity analysis within the genus. (b) Collinearity analysis across genera.
Code availability
No custom code was used in this study. All bioinformatics tools, commands and pipelines used in data processing were executed following the manual and protocols provided by the respective software developers. The versions of the software used, along with their corresponding parameters, have been thoroughly described in the Methods section.
References
Sabetian, A. The Association of Physical and Environmental Factors with Abundance and Distribution Patterns of Groupers around Kolombangara Island, Solomon Islands. Environ. Biol. Fishes 68, 93–99, https://doi.org/10.1023/A:1026048115070 (2003).
Rimmer, M. A. & Glamuzina, B. A review of grouper (Family Serranidae: Subfamily Epinephelinae) aquaculture from a sustainability science perspective. Rev. Aquac. 11, 58–87, https://doi.org/10.1111/raq.12226 (2019).
FAO. The State of World Fisheries and Aquaculture 2022. Towards Blue Transformation. (Rome, FAO, 2022).
Fisheries Administration Bureau, M. o. A. China Fishery Statistics Yearbook (2023). (China Agriculture Press, 2023).
Cao, X. et al. Chromosome-Level Genome Assembly of the Speckled Blue Grouper (Epinephelus cyanopodus) Provides Insight into Its Adaptive Evolution. Biology 11, 1810, https://doi.org/10.3390/biology11121810 (2022).
Ge, H. et al. De novo assembly of a chromosome-level reference genome of red-spotted grouper (Epinephelus akaara) using nanopore sequencing and Hi-C. Mol. Ecol. Resour. 19, 1461–1469, https://doi.org/10.1111/1755-0998.13064 (2019).
Li, S. et al. Mechanisms of sex differentiation and sex reversal in hermaphrodite fish as revealed by the Epinephelus coioides genome. Mol. Ecol. Resour. 23, 920–932, https://doi.org/10.1111/1755-0998.13753 (2023).
Wang, L. et al. A chromosome-level genome assembly of the potato grouper (Epinephelus tukula). Genomics 114, 110473, https://doi.org/10.1016/j.ygeno.2022.110473 (2022).
Yang, Y. et al. Whole-genome sequencing of brown-marbled grouper (Epinephelus fuscoguttatus) provides insights into adaptive evolution and growth differences. Mol. Ecol. Resour. 22, 711–723, https://doi.org/10.1111/1755-0998.13494 (2022).
Zhou, Q., Gao, H., Xu, H., Lin, H. & Chen, S. A Chromosomal-scale Reference Genome of the Kelp Grouper Epinephelus moara. Mar Biotechnol 23, 12–16, https://doi.org/10.1007/s10126-020-10003-6 (2021).
Zhou, Q. et al. A chromosome-level genome assembly of the giant grouper (Epinephelus lanceolatus) provides insights into its innate immunity and rapid growth. Mol. Ecol. Resour. 19, 1322–1332, https://doi.org/10.1111/1755-0998.13048 (2019).
Wang, D. et al. Whole Genome Sequencing of the Giant Grouper (Epinephelus lanceolatus) and High-Throughput Screening of Putative Antimicrobial Peptide Genes. Mar. Drugs 17, 503, https://doi.org/10.3390/md17090503 (2019).
Yang, Y. et al. Assembly of Genome and Resequencing Provide Insights into Genetic Differentiation between Parents of Hulong Hybrid Grouper (Epinephelus fuscoguttatus ♀ × E. lanceolatus ♂). Int J Mol Sci. 24, 12007, https://doi.org/10.3390/ijms241512007 (2023).
Han, W. et al. Improved chromosomal-level genome assembly and re-annotation of leopard coral grouper. Sci. Data 10, 156, https://doi.org/10.1038/s41597-023-02051-z (2023).
Wang, Y. et al. Chromosome Genome Assembly of the Leopard Coral Grouper (Plectropomus leopardus) With Nanopore and Hi-C Sequencing Data. Front Genet. 11, https://doi.org/10.3389/fgene.2020.00876 (2020).
Yang, Y. et al. Whole-genome sequencing of leopard coral grouper (Plectropomus leopardus) and exploration of regulation mechanism of skin color and adaptive evolution. Zool. Res. 41, 328, https://doi.org/10.24272/j.issn.2095-8137.2020.038 (2020).
Zhou, Q. et al. De novo sequencing and chromosomal-scale genome assembly of leopard coral grouper, Plectropomus leopardus. Mol. Ecol. Resour. 20, 1403–1413, https://doi.org/10.1111/1755-0998.13207 (2020).
Yang, Y. et al. Chromosome Genome Assembly of Cromileptes altivelis Reveals Loss of Genome Fragment in Cromileptes Compared with Epinephelus Species. Genes 12, 1873, https://doi.org/10.3390/genes12121873 (2021).
Xie, Z. et al. Chromosome-Level Genome Assembly and Transcriptome Comparison Analysis of Cephalopholis sonnerati and Its Related Grouper Species. Biology 11, 1053, https://doi.org/10.3390/biology11071053 (2022).
Ma, K. Y., Craig, M. T., Choat, J. H. & van Herwerden, L. The historical biogeography of groupers: Clade diversification patterns and processes. Mol. Phylogenet. Evol. 100, 21–30, https://doi.org/10.1016/j.ympev.2016.02.012 (2016).
Zhang, W. et al. The genetic mechanism of body size variation in groupers: insights from phylotranscriptomics. Zool Res. https://doi.org/10.24272/j.issn.2095-8137.2023.222 (2024).
Craig, M. T., Sadovy de Mitcheson, Y. & Heemstra, P. C. Groupers of the World: A Field and Market Guide. (2011).
Liu, M. et al. Primary male development of two sequentially hermaphroditic groupers, Epinephelus akaara and Epinephelus awoara (Perciformes: Epinephelidae). J. Fish Biol. 88, 1598–1613, https://doi.org/10.1111/jfb.12936 (2016).
Li, Z. et al. The complete mitochondrial genome of the hybrid offspring Epinephelus awoara ♀ × Epinephelus tukula ♂. Mitochondrial DNA B Resour 5, 1025–1026, https://doi.org/10.1080/23802359.2020.1721356 (2020).
Chen, B. et al. Biology and hatchery of Epinephelus awoara. Hebei Fisheries 2, 29–31 (2006).
Wang, S. et al. Characterization of yellow grouper Epinephelus awoara (Serranidae) karyotype by chromosome bandings and fluorescence in situ hybridization. J. Fish Biol. 80, 866–875, https://doi.org/10.1111/j.1095-8649.2012.03230.x (2012).
Yang, K. et al. Genetic Structure and Demographic History of Yellow Grouper (Epinephelus awoara) from the Coast of Southeastern Mainland China, Inferred by Mitochondrial, Nuclear and Microsatellite DNA Markers. Diversity 14, 439, https://doi.org/10.3390/d14060439 (2022).
Zhao, L. et al. Twelve novel polymorphic microsatellite loci for the Yellow grouper (Epinephelus awoara) and cross-species amplifications. Curr. Biol. 10, 743–745, https://doi.org/10.1007/s10592-008-9635-9 (2009).
Qu, M. et al. Complete mitochondrial genome of yellow grouper Epinephelus awoara (Perciformes, Epinephelidae). Mitochondrial DNA 23, 432–434, https://doi.org/10.3109/19401736.2012.710217 (2012).
Gong, G. et al. A chromosome-level genome assembly of the darkbarbel catfish Pelteobagrus vachelli. Sci. Data 10, 598, https://doi.org/10.1038/s41597-023-02509-0 (2023).
Zhou, Z. et al. The sequence and de novo assembly of Takifugu bimaculatus genome using PacBio and Hi-C technologies. Sci. Data 6, 187, https://doi.org/10.1038/s41597-019-0195-2 (2019).
Yekefenhazi, D. et al. Chromosome-level genome assembly of Nibea coibor using PacBio HiFi reads and Hi-C technologies. Sci. Data 9, 670, https://doi.org/10.1038/s41597-022-01804-6 (2022).
Eid, J. et al. Real-Time DNA Sequencing from Single Polymerase Molecules. Science 323, 133–138, https://doi.org/10.1126/science.1162986 (2009).
Rao, S. S. P. et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159, 1665–1680, https://doi.org/10.1016/j.cell.2014.11.021 (2014).
Oikonomopoulos, S. et al. Methodologies for Transcript Profiling Using Long-Read Technologies. Front. Genet. 11, https://doi.org/10.3389/fgene.2020.00606 (2020).
Zhao, L. et al. Analysis of Transcriptome and Epitranscriptome in Plants Using PacBio Iso-Seq and Nanopore-Based Direct RNA Sequencing. Front. Genet. 10, 253, https://doi.org/10.3389/fgene.2019.00253 (2019).
Gonzalez-Garay, M. L. in Transcriptomics and Gene Regulation (ed Jiaqian, Wu) 141–160 (Springer Netherlands, 2016).
Chen, S. et al. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinformatics/bty560 (2018).
Deorowicz, S. et al. KMC 2: fast and resource-frugal k-mer counting. Bioinformatics 31, 1569–1576, https://doi.org/10.1093/bioinformatics/btv022 (2015).
Sun, H. et al. findGSE: estimating genome size variation within human and Arabidopsis using k-mer frequencies. Bioinformatics 34, 550–557, https://doi.org/10.1093/bioinformatics/btx637 (2018).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33, 2202–2204, https://doi.org/10.1093/bioinformatics/btx153 (2017).
Cheng, H. et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175, https://doi.org/10.1038/s41592-020-01056-5 (2021).
Hu, J. et al. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255, https://doi.org/10.1093/bioinformatics/btz891 (2020).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595, https://doi.org/10.1093/bioinformatics/btp698 (2010).
Li, H. Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110, https://doi.org/10.1093/bioinformatics/btw152 (2016).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, https://doi.org/10.1093/bioinformatics/btp352 (2009).
Danecek, P. & McCarthy, S. A. BCFtools/csq: haplotype-aware variant consequences. Bioinformatics 33, 2037–2039, https://doi.org/10.1093/bioinformatics/btx100 (2017).
Chaisson, M. J. & Tesler, G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics 13, 238, https://doi.org/10.1186/1471-2105-13-238 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359, https://doi.org/10.1038/nmeth.1923 (2012).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259, https://doi.org/10.1186/s13059-015-0831-x (2015).
Burton, J. N. et al. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125, https://doi.org/10.1038/nbt.2727 (2013).
Wang, X. & Wang, L. GMATA: An Integrated Software Package for Genome-Scale SSR Mining, Marker Development and Viewing. Front. Plant Sci. 7, 1350, https://doi.org/10.3389/fpls.2016.01350 (2016).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580, https://doi.org/10.1093/nar/27.2.573 (1999).
Han, Y. & Wessler, S. R. MITE-Hunter: a program for discovering miniature inverted-repeat transposable elements from genomic sequences. Nucleic Acids Res. 38, e199–e199, https://doi.org/10.1093/nar/gkq862 (2010).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proceedings of the National Academy of Sciences 117, 9451–9457, https://doi.org/10.1073/pnas.1921046117 (2020).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268, https://doi.org/10.1093/nar/gkm286 (2007).
Ellinghaus, D., Kurtz, S. & Willhoeft, U. LTRharvest, an efficient and flexible software for de novo detection of LTR retrotransposons. BMC Bioinformatics 9, 18, https://doi.org/10.1186/1471-2105-9-18 (2008).
Ou, S. & Jiang, N. LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 176, 1410–1422, https://doi.org/10.1104/pp.17.01310 (2018).
Abrusán, G., Grundmann, N., DeMester, L. & Makalowski, W. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics 25, 1329–1330, https://doi.org/10.1093/bioinformatics/btp084 (2009).
Bedell, J. A., Korf, I. & Gish, W. MaskerAid: a performance enhancement to RepeatMasker. Bioinformatics 16, 1040–1041, https://doi.org/10.1093/bioinformatics/16.11.1040 (2000).
Keilwagen, J. et al. Using intron position conservation for homology-based gene prediction. Nucleic Acids Res. 44, e89–e89, https://doi.org/10.1093/nar/gkw092 (2016).
Zebrafish Genome Assembly GRCz11 Statistics, Genome Reference Consortium. https://www.ncbi.nlm.nih.gov/grc/zebrafish/data. (2018).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21, https://doi.org/10.1093/bioinformatics/bts635 (2013).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Bio. 20, 278, https://doi.org/10.1186/s13059-019-1910-1 (2019).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7, https://doi.org/10.1186/gb-2008-9-1-r7 (2008).
Stanke, M. et al. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 24, 637–644, https://doi.org/10.1093/bioinformatics/btn013 (2008).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence data bank and its supplement TrEMBL in 1999. Nucleic Acids Res. 27, 49–54, https://doi.org/10.1093/nar/27.1.49 (1999).
Ogata, H. et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 27, 29–34, https://doi.org/10.1093/nar/27.1.29 (1999).
Galperin, M. Y. et al. Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269, https://doi.org/10.1093/nar/gku1223 (2015).
Ashburner, M. et al. Gene Ontology: tool for the unification of biology. Nat. Genet. 25, 25–29, https://doi.org/10.1038/75556 (2000).
Chan, P. P. et al. tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096, https://doi.org/10.1093/nar/gkab688 (2021).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935, https://doi.org/10.1093/bioinformatics/btt509 (2013).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124, https://doi.org/10.1093/nar/gki081 (2005).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108, https://doi.org/10.1093/nar/gkm160 (2007).
Chen, T. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genomics Proteomics Bioinformatics 19, 578–583, https://doi.org/10.1016/j.gpb.2021.08.001 (2021).
Members, C. N. & Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2023. Nucleic Acids Res. 51, D18–D28, https://doi.org/10.1093/nar/gkac1073 (2023).
NGDC Genome Sequence Archive https://bigd.big.ac.cn/gsa/browse/CRA013097 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP479893 (2023).
NCBI GenBank https://identifiers.org/ncbi/insdc.gca:GCA_035609425.1 (2024).
Chen, M. et al. Genome Warehouse: A Public Repository Housing Genome-scale Data. Genomics Proteomics Bioinformatics 19, 584–589, https://doi.org/10.1016/j.gpb.2021.04.001 (2021).
NGDC Genome Warehouse https://ngdc.cncb.ac.cn/gwh/Assembly/82944/show (2023).
Rhie, A. et al. Merqury: reference-free quality, completeness, and phasing assessment for genome assemblies. Genome Biology 21, 245, https://doi.org/10.1186/s13059-020-02134-9 (2020).
Parra, G. et al. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067, https://doi.org/10.1093/bioinformatics/btm071 (2007).
Manni, M. et al. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 38, 4647–4654, https://doi.org/10.1093/molbev/msab199 (2021).
Tang, H. et al. Synteny and Collinearity in Plant Genomes. Science 320, 486–488, https://doi.org/10.1126/science.1153917 (2008).
Acknowledgements
This work is supported by National Natural Science Foundation of China (32273136, U22A20531), China Agriculture Research System of MOF and MARA (CARS-47), the Science and Technology Planning Project of Guangdong Province (2023B1212060023) and Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai) (No. SML2023SP201). We also thank GrandOmics Technologies (Wuhan, China) for their invaluable technical support in this study.
Author information
Authors and Affiliations
Contributions
Z.M. and W.Z. conceived and designed the study. Z.M. and X.L. coordinated and supervised the whole study. W.Z. and Y.Y. conducted the genome assembly and bioinformatics analysis. S.H. and Q.R. prepared the samples and the figures. W.Z. drafted the manuscript. D.L., L.W., X.W. and X.W. participated in discussions and provided suggestions for manuscript improvement. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, W., Yang, Y., Hua, S. et al. Chromosome-level genome assembly and annotation of the yellow grouper, Epinephelus awoara. Sci Data 11, 151 (2024). https://doi.org/10.1038/s41597-024-02989-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-02989-8
